
Применяемые в качестве стартовых режимов современные схемы антиретровирусной терапии (АРТ), как правило, содержат 2 препарата из группы нуклеозидных (нуклеотидных) ингибиторов обратной транскриптазы ВИЧ (НИОТ/НтИОТ), в сочетании с ненуклеозидным ингибитором обратной транскриптазы ВИЧ (ННИОТ), либо ингибитором протеазы или интегразы ВИЧ.
Комбинация, состоящая из тенофовира/эмтрицитабина/эфавиренза (TDF/FTC/EFV), была первым режимом, который применяли в виде 1 таблетки 1 раз в сутки. В настоящее время сочетание препаратов тенофовир/эмтрицитабин/рилпивирин (TDF/FTC/RPV) является единственной зарегистрированной в России схемой АРТ с приемом 1 таблетки 1 раз в сутки [1]. Вместе с тем на основании проведенных исследований комбинация TDF/FTC/RPV рекомендована в качестве режима первой линии (пациентам, ранее не получавшим лечения) больным с исходным уровнем РНК ВИЧ < 100 000 копий/мл, поскольку при более высоких уровнях вирусной нагрузки эффективность терапии по этой схеме может быть недостаточна [2–5]. Применение в режимах АРТ первой линии препарата EFV более чем у 60% пациентов сопровождается развитием нежелательных явлений (НЯ) со стороны центральной нервной системы, в результате чего схема TDF/FTC/EFV в рекомендациях специалистов США была переведена из приоритетных в альтернативные [3].
В августе 2014 г. было начато исследование № 219 (II–III фаза) – международное многоцентровое рандомизированное частично слепое клиническое исследование эффективности, безопасности и подбора оптимальной дозировки препарата элпивирин (VM-1500; Elpida®) в сравнении с EFV на фоне стандартной базисной АРТ, состоящей из 2 НИОТ/НтИОТ, у инфицированных ВИЧ-1 пациентов, ранее не получавших лечения [6].
 Исследование включало 2 этапа. Основной целью 1-го этапа был выбор оптимальной дозы элпивирина (20 или 40 мг в сутки) в сочетании с базисной терапией (2 НИОТ/НтИОТ) на основании эффективности (доля больных с РНК ВИЧ < 400 копий/мл через 12 недель лечения) в сравнении с EFV.
Исследование включало 2 этапа. Основной целью 1-го этапа был выбор оптимальной дозы элпивирина (20 или 40 мг в сутки) в сочетании с базисной терапией (2 НИОТ/НтИОТ) на основании эффективности (доля больных с РНК ВИЧ < 400 копий/мл через 12 недель лечения) в сравнении с EFV.
На 1-м этапе в исследование было включено 90 пациентов с ВИЧ-1-инфекцией, ранее не получавших АРТ, которые были рандомизированы в 3 группы: в 1-й 30 больных получали элпивирин в дозе 20 мг в сутки; во 2-й 30 больных получали элпивирин по 40 мг в сутки; в 3-й (группа сравнения) 30 больных получали EFV по 600 мг в сутки. Кроме того, все пациенты получали 2 НИОТ/НтНИОТ – TDF/FTC [6].
Через 12 недель лечения, по завершении 1-го этапа исследования, были получены результаты, доказавшие равную эффективность элпивирина в дозах 20 мг и 40 мг в сочетании с TDF/FTC и схемы EFV + TDF/FTC вне зависимости от исходного уровня вирусной нагрузки.
Наиболее безопасным режимом после 12 недель терапии оказалась схема, включавшая суточную дозу препарата 20 мг. Особо были проанализированы нарушения со стороны ЦНС и психической деятельности (так называемые НЯ «особого интереса»), которые достоверно реже наблюдали у пациентов 1-й группы (26,7%) по сравнению с пациентами 3-й группы (57,1%) [6].
На основании результатов, полученных на 1-м этапе, для проведения дальнейших исследований была одобрена доза элпивирина 20 мг в сутки.
Основной целью 2-го этапа исследования было сравнение эффективности (доля больных с РНК ВИЧ < 50 копий/мл через 24 недели лечения) выбранной дозы элпивирина (20 мг в сутки) и EFV в сочетании с 2 НИОТ/НтИОТ.
Помимо целей, которые были решены на 1-м этапе, в исследовании был сформулирован ряд дополнительных целей:
- вирусологических – среднее снижение РНК ВИЧ в течение 48 недель исследуемой терапии; доля пациентов, продолживших прием исследуемой терапии до 48 недели;
- иммунологических – изменение абсолютного числа СD4+- и СD8+-лимфоцитов в течение 48 недель терапии;
- оценка резистентности ВИЧ – доля пациентов, у которых в течение 48 недель развилась резистентность ВИЧ-1 к исследуемой терапии;
- оценка безопасности – частота развития НЯ различной степени тяжести по данным субъективных жалоб, физикального осмотра, жизненных показателей, лабораторных исследований; частота развития НЯ «особого интереса», в том числе нарушений со стороны ЦНС.
Материалы и методы
В исследование включали мужчин и женщин в возрасте 18 лет и старше с серологически подтвержденной ВИЧ-1-инфекцией (методами ИФА и иммунного блотинга), стабильным клиническим течением ВИЧ-инфекции (клинические стадии 1 или 2 по классификации ВОЗ), которым по решению Исследователя показано начало АРТ [согласно Сводному руководству ВОЗ по использованию антиретровирусных препаратов (АРП) для лечения и профилактики ВИЧ-инфекции от 2013 г.], с уровнем РНК ВИЧ-1 в плазме крови ≥ 5000 копий/мл и количеством СD4+-лимфоцитов > 200 клеток/мкл на скрининге. Перед проведеним скрининга все пациенты подписывали информационный листок пациента и форму информированного согласия на участие в исследовании. Кроме того, было получено согласие пациентов на использование в течение всего исследования адекватных методов контрацепции (презерватив со спермицидом).
 На 2-м этапе в исследовании приняли участие 120 больных ВИЧ-1-инфекцией, ранее не получавших АРТ. В основную группу были включены 30 больных, завершивших 1-й этап исследования и получавших выбранную дозу элпивирина (20 мг), а также дополнительно рандомизированы 30 новых пациентов с ВИЧ-инфекцией, соответствующих критериям включения. Контрольная группа состояла из 30 больных, получавших на 1-м этапе исследования EFV (600 мг в сутки) и продолживших терапию. В эту группу также было дополнительно рандомизировано 30 новых пациентов в соответствии с критериями включения.
На 2-м этапе в исследовании приняли участие 120 больных ВИЧ-1-инфекцией, ранее не получавших АРТ. В основную группу были включены 30 больных, завершивших 1-й этап исследования и получавших выбранную дозу элпивирина (20 мг), а также дополнительно рандомизированы 30 новых пациентов с ВИЧ-инфекцией, соответствующих критериям включения. Контрольная группа состояла из 30 больных, получавших на 1-м этапе исследования EFV (600 мг в сутки) и продолживших терапию. В эту группу также было дополнительно рандомизировано 30 новых пациентов в соответствии с критериями включения.
Таким образом, через 24 недели исследования (2-й этап) сравнивали результаты, полученные от пациентов основной и контрольной групп, каждая из которых на момент начала исследования включала 60 больных ВИЧ-инфекцией.
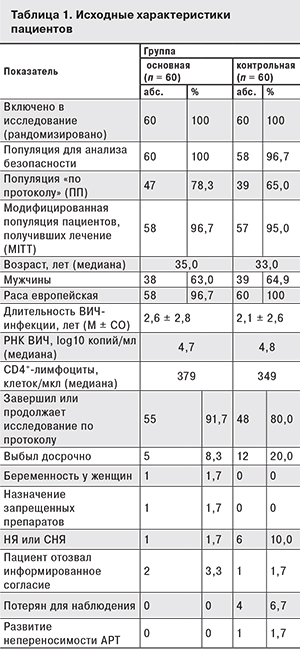
Исходные характеристики пациентов представлены в табл. 1. Существенных различий между группами пациентов в исходных показателях обнаружено не было. Среди вторичных заболеваний, имевших место в анамнезе, у 1,8–5,2% пациентов отмечали пневмонии, у 6,9–18% – кандидоз слизистых оболочек полости рта, у 1,7–7,0% – опоясывающий лишай, у 3,4–7,0% – эрозии шейки матки, у 3,4–3,5% – анемию.
Запланированная длительность участия пациентов в исследовании составляет 54 недели (2 недели – скрининг, 48 недель – исследуемая терапия и 4 недели – наблюдение). Набор пациентов в исследование начат в январе 2014 г., окончание исследования ожидается в ноябре 2016 г.
На 2-м этапе в исследовании приняли участие сотрудники центров по профилактике и борьбе со СПИДом Республики Татарстан и Удмуртской Республики, Пермского края, Волгоградской, Липецкой, Московской, Рязанской и Самарской областей, Москвы и Санкт-Петербурга, а также специалисты Федерального Центра по профилактике и борьбе со СПИДом (Москва).
На данном этапе проведен промежуточный (через 24–48 недель лечения) анализ эффективности и безопасности АРТ.
Абсолютное и относительное содержание CD4+- и CD8+-лимфоцитов (определяли методом проточной цитометрии) и уровень РНК ВИЧ (ПЦР) исследовали на скрининге через 4, 8, 12, 24, 36 и 48 недель АРТ. Параметры анализа периферической крови и биохимического анализа крови – до лечения, через 4, 8, 12, 24, 36 и 48 недель терапии.
 Для оценки эффективности терапии оценивали модифицированную популяцию пациентов, получивших лечение, и популяцию пациентов «по протоколу». Модифицированная популяция пациентов, получивших лечение (MITT – modified intent-to-treat), соответствовала всем рандомизированным пациентам, которые получили, по крайней мере, одну дозу исследуемого препарата или препарата сравнения и у которых имелось хотя бы одно измерение вирусной нагрузки ВИЧ-1 после исходного. Популяция MITT являлась основной для анализа. Популяции «по протоколу» (ПП-анализ) соответствовали все пациенты популяции MITT, которые полностью завершили исследование (или его этап), имели оценки для первичного анализа эффективности и считались комплаентными. Комплаентными считали пациентов, не имевших каких-либо серьезных нарушений протокола в ходе исследования.
Для оценки эффективности терапии оценивали модифицированную популяцию пациентов, получивших лечение, и популяцию пациентов «по протоколу». Модифицированная популяция пациентов, получивших лечение (MITT – modified intent-to-treat), соответствовала всем рандомизированным пациентам, которые получили, по крайней мере, одну дозу исследуемого препарата или препарата сравнения и у которых имелось хотя бы одно измерение вирусной нагрузки ВИЧ-1 после исходного. Популяция MITT являлась основной для анализа. Популяции «по протоколу» (ПП-анализ) соответствовали все пациенты популяции MITT, которые полностью завершили исследование (или его этап), имели оценки для первичного анализа эффективности и считались комплаентными. Комплаентными считали пациентов, не имевших каких-либо серьезных нарушений протокола в ходе исследования.
Описательную статистику по вирусной нагрузке представляли на обычной шкале (десятичной) и логарифмической (десятичный логарифм). Для сравнения групп терапии по изменению вирусной нагрузки была построена смешанная линейная модель.
Изменение РНК ВИЧ и количества CD4+-лимфоцитов описывали по временным точкам (визитам) исследования как непрерывные величины. Для каждой временной точки (исключая исходный уровень) вычисляли изменение параметра относительно исходного уровня и представляли в таблицах описательной статистикой. Внутригрупповые изменения параметра тестировали с помощью t-теста Стьюдента (для нормально распределенных данных) или знакового критерия Вилкоксона (Манна–Уитни) (для данных, не имеющих нормального распределения). В качестве теста на нормальность распределения использовали тест Шапиро–Вилка.
 Регистрацию НЯ проводили с момента подписания пациентом формы информированного согласия и до 30 дней после последнего визита пациента в исследовательский центр или проведения последней процедуры, предусмотренной протоколом. Все НЯ, зарегистрированные в индивидуальной регистрационной карте пациента, были разделены на явления, возникшие в ходе исследования, и все остальные. Учитывали всех пациентов, которые получили, по крайней мере, одну дозу исследуемого препарата или препарата сравнения. Сравнение было проведено для НЯ «особого интереса», развившихся в ходе исследования, к которым относили НЯ со стороны ЦНС и некоторые другие. До начала анализа НЯ как явления «особого интереса» квалифицировали специалисты спонсора. Группы больных сравнивали по частоте встречаемости данных НЯ (рассматривали число пациентов с НЯ «особого интереса»). Этот параметр представляли в абсолютных и относительных значениях.
Регистрацию НЯ проводили с момента подписания пациентом формы информированного согласия и до 30 дней после последнего визита пациента в исследовательский центр или проведения последней процедуры, предусмотренной протоколом. Все НЯ, зарегистрированные в индивидуальной регистрационной карте пациента, были разделены на явления, возникшие в ходе исследования, и все остальные. Учитывали всех пациентов, которые получили, по крайней мере, одну дозу исследуемого препарата или препарата сравнения. Сравнение было проведено для НЯ «особого интереса», развившихся в ходе исследования, к которым относили НЯ со стороны ЦНС и некоторые другие. До начала анализа НЯ как явления «особого интереса» квалифицировали специалисты спонсора. Группы больных сравнивали по частоте встречаемости данных НЯ (рассматривали число пациентов с НЯ «особого интереса»). Этот параметр представляли в абсолютных и относительных значениях.
Результаты
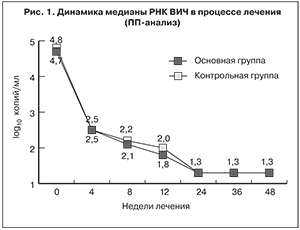
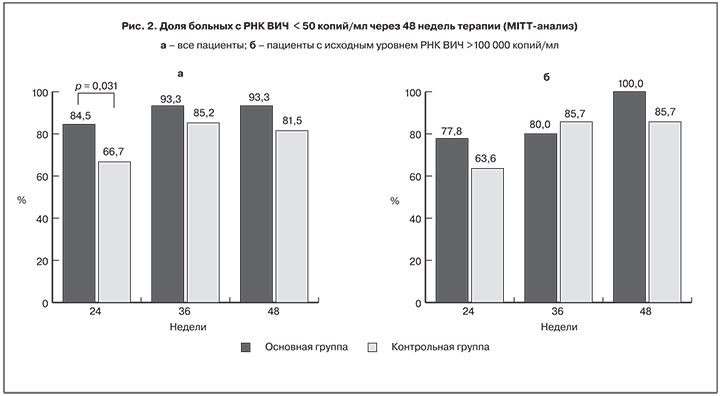
Через 24 недели АРТ медиана РНК ВИЧ-1 была равна 1,3 log10 копий/мл у пациентов обеих групп (p < 0,001 по сравнению с исходными значениями) и сохранялась на этом уровне через 36–48 недель исследования (рис. 1). Спустя 24 недели терапии доля больных с уровнем РНК ВИЧ-1 < 50 копий/мл составила: MITT-анализ – 84,5 и 66,7% (р = 0,031), ПП-анализ – 85,1 и 74,1% соответственно. Анализ доли пациентов с неопределяемым уровнем РНК ВИЧ через 24 недель АРТ при исходно высоком (> 100 000 копий/мл) уровне вирусной нагрузки не выявил достоверных различий между пациентами обеих групп. Спустя 36 и 48 недель исследования доля больных с вирусной нагрузой ниже порога определения тест-системой была сопоставима у пациентов обеих групп (рис. 2).
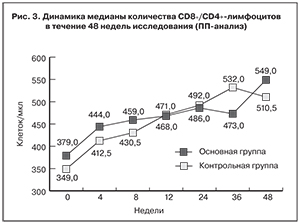
Динамика медианы количества CD4+-лимфоцитов представлена на рис. 3. Через 24 недели АРТ медиана количества CD4+-лимфоцитов составляла 468 и 471 клетку/мкл, а к 48 неделям лечения – 549 и 510,5 клеток/мкл соответственно. Среднее количество CD4+-лимфоцитов у больных основной группы через 24 недели терапии увеличилось с 398,5 ± 161,9 до 540,3 ± 215,2 клеток/мкл, а в контрольной – с 419,6 ± 196,8 до 532,0 ± 199,5 клеток/мкл (p < 0,001 для обеих групп по сравнению с показателями до лечения). Спустя 48 недель исследования прирост среднего количества СD4+-лимфоцитов составил 190 и 147,4 клетки/мкл соответственно. У пациентов обеих групп через 24 недели лечения отметили существенное уменьшение медианы количества CD8+-лимфоцитов: в основной группе – с 919 до 839 клеток/мкл, в контрольной – с 1038 до 938 клеток/мкл. Снижение количества CD8+-лимфоцитов сохранялось через 36 и 48 недель терапии, оно в совокупности с увеличением количества CD4+-лимфоцитов обусловило увеличение медианы иммунорегуляторного индекса (соотношение CD4+/CD8+-лимфоцитов) с 0,412 и 0,336 до лечения до 0,579 и 0,525 после 24 недель АРТ.
Таким образом, предварительные результаты исследования свидетельствуют о сопоставимой эффективности схем АРТ, включавших элпивирин в суточной дозировке 20 мг, и схемы, содержавшей EFV. Вместе с тем при проведении MITT-анализа через 24 недели терапии было показано, что в основной группе доля больных с уровнем РНК ВИЧ < 50 копий/мл была на 17,8% выше, чем в контрольной. Иммунологическая эффективность обеих схем АРТ также была сходной, однако несколько большее снижение количества CD8+-лимфоцитов у пациентов основной группы обусловило увеличение иммунорегуляторного индекса на 0,47, тогда как в контрольной группе – только на 0,26.
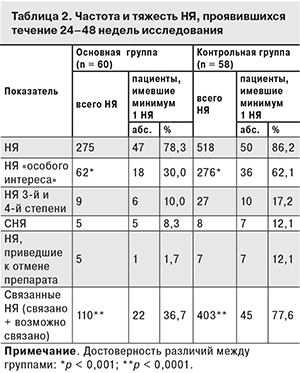
НЯ любой степени тяжести были зарегистрированы у 78,3 и 86,2% больных соответственно. В том числе НЯ тяжелой (3-й и 4-й) степени были отмечены у 10 и 17,2% пациентов. НЯ, связанные или возможно связанные с лечением, достоверно чаще регистрировали у больных контрольной группы (р < 0,0001; табл. 2).
У 12 пациентов в процессе исследования были зарегистрированы серьезные НЯ (СНЯ). Из 5 пациентов основной группы у 3 имели место СНЯ легкой и умеренной степени тяжести (пневмония нижней доли правого легкого после переохлаждения, конкремент в средней почечной лоханке справа, инфильтративный туберкулез левого легкого в фазе распада, МБТ+) и у 1 – тяжелой степени (повышение уровня КФК). В 1 случае была зафиксирована тяжелая черепно-мозговая травма, закончившаяся летальным исходом (несчастный случай – автоавария на пешеходном переходе). Во всех случаях связь СНЯ с исследуемым препаратом не установлена.
В контрольной группе наблюдали 7 случаев СНЯ. 3 СНЯ умеренной степени тяжести (пневмония, аденовирусная инфекция и травма мениска) не были связаны с АРТ, 4 СНЯ тяжелой степени (цитолитический синдром, реакция гиперчувствительности, кожно-аллергическая реакция), возможно/вероятно, связаны с приемом EFV.
Во всех случаях СНЯ были купированы. У 1 пациента элпивирин был отменен вследствие наступления летального исхода, в остальных случаях СНЯ препарат не отменяли и не изменяли его дозу. У 7 больных контрольной группы при развитии СНЯ схему АРТ изменяли (была произведена замена EFV) (см. табл. 2).
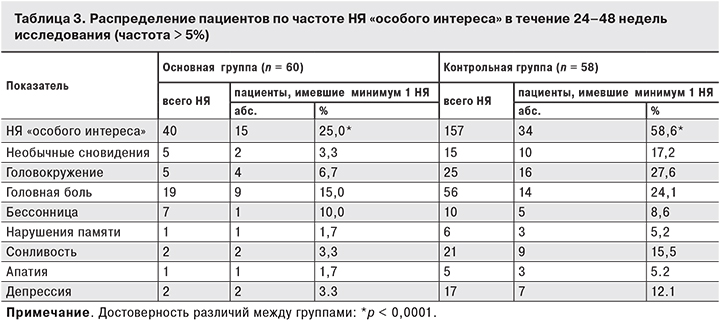
В соответствии с протоколом исследования были выделены НЯ «особого интереса», которые имели место у 30% больных основной и у 62,1% больных контрольной группы (р < 0,001). Нарушения со стороны ЦНС были выявлены у 25 и 58,6% больных соответственно (p < 0,001; табл. 3). При этом НЯ тяжелой степени были отмечены только у 2 (1,7%) пациентов основной группы, тогда как в контрольной группе их обнаруживали в 5 (8,6%) случаях.
У 5 больных основной группы наблюдали 14 НЯ, связанных с приемом препарата, 7 из них были со стороны ЦНС и психической деятельности: головная боль, головокружение, чувство тревожности, нарушения со стороны вегетативной нервной системы. Среди других НЯ, связанных или возможно связанных с приемом элпивирина, регистрировали аутоиммунный тиреоидит, диарею, тошноту, астению, боль в груди, повышение уровня КФК и одышку. У больных контрольной группы развитие НЯ было связано с приемом EFV у 30 (51,7%) больных. Всего зарегистрировано 172 случая НЯ, среди которых 121 был связан с нарушениями со стороны ЦНС и психической деятельности: необычные сновидения, угнетенный уровень сознания, снижение двигательной активности, нарушение внимания, памяти, головная боль, головокружение, бессонница, сонливость, тревожность, раздражительность, депрессия, панические реакции и др.
Среди неврологических НЯ, частота регистрации которых составляла 5% и более, пациенты основной группы отметили только головную боль и головокружение, а пациентов контрольной группы – необычные сновидения, головокружение, головную боль, бессонницу, нарушения сна, сонливость, нарушения памяти, апатию и депрессию (см. табл. 3).
НЯ со стороны ЖКТ в основной группе регистрировали у 18,3% больных, в контрольной – у 27,6%. Диарею легкой и средней тяжести отмечали у 6,7% пациентов основной группы, легкой степени тяжести – у 5,2% пациентов контрольной группы; тошноту легкой степени тяжести – у 5,0 и 10,3% соответственно.
Развитие НЯ со стороны кожных покровов (акне, выпадение волос, папулезная сыпь, себорейный дерматит) наблюдали только у 4 (6,7%) пациентов основной группы, при этом выявленные НЯ не были связаны с АРТ. Среди пациентов контрольной группы подобные НЯ регистрировали у 19 (32,8%) больных. Связь НЯ с приемом EFV была отмечена в 44,4% случаев.
Оценка изменений показателей периферической крови показала, что наиболее частыми НЯ в течение первых 24 недель лечения были лейкопения и нейтропения. Развитие лейкопении легкой степени тяжести регистрировали в основной группе у 28,3% пациентов, в контрольной – у 20,7%. Нейтропению легкой степени тяжести отмечали у 13,3 и у 6,9% пациентов соответственно.
Поскольку в исследование не включали больных, страдающих вирусными гепатитами, исходный уровень АлАТ был в пределах нормы у 86,7% больных основной группы и у 93,3% больных контрольной группы, уровень АсАТ – у 91,4 и 94,8% пациентов соответственно. В течение 24 недель исследования только у 1,7% пациентов обеих групп регистрировали повышение уровней АлАТ и/или АсАТ легкой степени тяжести, возможно связанное с принимаемыми препаратами. Повышение уровня ГГТ имело место у 11 (18,9%) больных основной группы (у 6 пациентов оно, возможно, было связано с лечением) и у 6 (10,3%) больных контрольной группы (в 5 случаях, возможно, связанное с приемом EFV). У 5 (8,3%) больных основной и у 4 (6,7%) больных контрольной группы наблюдали повышенный уровень КФК. Только у 2 пациентов (по 1 в каждой группе) это НЯ было тяжелой степени, причем у больного основой группы оно маловероятно было связано с приемом элпивирина, у больного контрольной группы – возможно связано с приемом EFV. У обоих пациентов это НЯ успешно разрешилось. Другие показатели биохимического анализа крови (содержание креатинина, мочевины, альбумина, ЛДГ, холестерина, глюкозы) изменялись незначительно. Ни в одном случае коррекции параметров не потребовалось.
Таким образом, схема АРТ, содержавшая элпивирин в дозе 20 мг в сутки в комбинации с TDF/FTC, через 24–48 недель лечения была не менее эффективна, чем схема EFV + TDF/FTC, вне зависимости от исходного уровня вирусной нагрузки. Через 24 недели АРТ медиана уровня РНК ВИЧ снизилась до 1,3 lg копий/мл и сохранялась на этом уровне до 48 недель исследования. Уже через 24 недели терапии в основной группе доля пациентов с уровнем РНК ВИЧ < 50 копий/мл была максимальной (MITT-анализ) и составила 84,5% (p = 0,031 с контролем). Иммунологическая эффективность обоих режимов лечения была неплохой, поскольку через 48 недель терапии прирост количества CD4+-лимфоцитов (по медиане) составил 170 и 161,5 клеток/мкл соответственно. Увеличение количества CD4+-лимфоцитов и снижение количества CD8+-лимфоцитов обусловило повышение значений иммунорегуляторного индекса (соотношение CD4+/CD8+-лимфоцитов), в большей степени отмеченное у пациентов основной группы (+0,47).
Безопасность применения в течение 24–48 недель схемы АРТ, включавшей элпивирин, была выше, чем при использовании схемы, содержавшей EFV. В основной группе пациентов частота НЯ, связанных или возможно связанных с применяемыми препаратами, была существенно ниже (36,7%) по сравнению с пациентами контрольной группы (77,6%; р < 0,0001).
Лишь у 12 пациентов (у 5 – в основной группе и у 7 – в контрольной) были зарегистрированы СНЯ. В основной группе ни у одного пациента не установлена их связь с приемом исследуемого препарата. Лишь в 1 случае лечение было прекращено из-за смерти больного в результате дорожно-транспортного происшествия. У всех больных контрольной группы при развитии СНЯ схема лечения была изменена.
Развитие НЯ «особого интереса» (нарушения со стороны ЦНС и психической деятельности) в основной группе выявляли существенно реже, чем в контрольной – у 30 и 62,1% больных соответственно (р < 0,001). Также реже у больных основной группы отмечали развитие НЯ со стороны ЖКТ и кожных покровов. В течение первых 24–48 недель исследования не было выявлено существенных изменений показателей биохимического анализа крови и параметров периферической крови, потребовавших отмены или изменения схемы лечения.


