
Разработка и внедрение в клиническую практику новых антиретровирусных препаратов (АРП) позволяет повысить эффективность лечения как у больных, ранее не получавших терапии, так и у больных с устойчивостью вируса к ряду лекарственных препаратов.
В настоящее время для лечения пациентов с ВИЧ-инфекцией в составе схем антиретровирусной терапии (АРТ) первой линии используют 3 препарата из группы ННИОТ – эфавиренз (EFV), рилпивирин (RPV) и невирапин (NVP) [1]. Препараты RPV и NVP рекомендуется назначать в составе альтернативных режимов АРТ первой линии [1–3]. Сравнительные исследования результатов применения препаратов RPV и EFV показали, что режимы АРТ, содержащие RPV, могут быть менее эффективными (по сравнению с EFV) у больных с исходным уровнем РНК ВИЧ > 100 000 копий/мл [4, 5]. При использовании NVP отмечено повышение частоты гепатотоксических реакций, в особенности у женщин с исходным количеством CD4+-лимфоцитов > 250 клеток/мкл [6]. Включение в состав схемы АРТ EFV сопровождается высокой (более 60%) частотой развития нежелательных явлений (НЯ) со стороны центральной нервной системы, в связи с чем одну из наиболее популярных до недавнего времени схем АРТ – тенофовир + эмтрицитабин + эфавиренз (TDF + FTC + EFV) – специалисты США в рекомендациях 2015 г. исключили из приоритетных схем терапии первой линии [3].
В период с 2010 по 2011 г. на базе Государственного научно-исследовательского центра профилактической медицины Росмедтехнологий было проведено исследование I фазы препарата VM-1500 по протоколу 01/HIV/2010 «Открытое клиническое исследование I фазы фармакокинетики и безопасности препарата VM-1500 в виде капсул при пероральном введении у здоровых добровольцев с перекрестным исследованием фармакокинетики в зависимости от приема пищи». Целью данного исследования было определение переносимости препарата, а также его фармакокинетических параметров при пероральном однократном приеме в диапазоне доз 10–80 мг.
В исследовании приняли участие 24 здоровых добровольца в составе 7 когорт. При однократном и многократном приеме VM-1500 здоровыми добровольцами серьезных НЯ зарегистрировано не было. Наблюдаемые НЯ были в основном легкой степени тяжести и ни в одном случае не потребовали медикаментозного лечения или досрочного выведения добровольцев из исследования.
Исследование эффективности и безопасности препарата VM-1500 (фаза IIa) было проведено у 16 больных ВИЧ-инфекцией, ранее не получавших АРТ, с исходным уровнем РНК ВИЧ > 5000 копий/мл и CD4+-лимфоцитов > 250 клеток/мкл [7]. 8 больных 1-й группы были рандомизированы в соотношении 7:1 (монотерапия VM-1500 в дозе 20 мг в сутки и плацебо). Через 7 дней лечения средний уровень РНК ВИЧ у пациентов, получавших VM-1500, снизился с 82 944 до 2133 копий/мл, а количество CD4+-лимфоцитов возросло с 551 до 592 клеток/мкл. У пациента, получавшего плацебо, не регистрировали существенной динамики уровня РНК ВИЧ и количества CD4+-лимфоцитов (303 000 и 248 000 копий/мл, 492 и 517 клеток/мкл соответственно).
Пациенты 2-й группы (8 человек) также были рандомизированы в соотношении 7:1 (монотерапия VM-1500 в дозе 40 мг в сутки и плацебо). Динамика изучаемых показателей у больных 2-й группы была аналогичной: при приеме VM-1500 уровень РНК ВИЧ снизился с 93 671 до 1316 копий/мл, количество CD4+-лимфоцитов возросло с 471 до 525 клеток/мкл. Показатели у пациента, получавшего плацебо, – 126 000–116 206 копий/мл и 424–388 клеток/мкл соответственно.
Таким образом, у больных ВИЧ-инфекцией, в течение 7 дней получавших монотерапию VM-1500, отмечено снижение уровня РНК ВИЧ в среднем на 1,8 log10 копий/мл.
 Терапия VM-1500 была безопасной. 3 пациента 1-й группы предъявляли жалобы на чувство легкой сухости во рту и полиурию, а 1 – на головную боль легкой степени выраженности. У пациентов 2-й группы каких-либо НЯ отмечено не было [7].
Терапия VM-1500 была безопасной. 3 пациента 1-й группы предъявляли жалобы на чувство легкой сухости во рту и полиурию, а 1 – на головную боль легкой степени выраженности. У пациентов 2-й группы каких-либо НЯ отмечено не было [7].
Проведенные исследования фаз I и IIа у здоровых добровольцев и пациентов с ВИЧ-1 при приеме VM-1500 в монорежиме в дозах 20 и 40 мг в течение 7 дней продемонстрировали значительный противовирусный эффект (медиана снижения РНК ВИЧ – 1,8 log10 копий/мл), высокую безопасность и хорошую переносимость препарата и явились основанием для изучения эффективности и безопасности применения VM-1500 в составе схемы АРТ.
Материалы и методы
Исследование HIV-VM1500-04 (фаза II) – многоцентровое, рандомизированное, частично слепое клиническое исследование эффективности, безопасности и подбора оптимальной дозировки препарата VM-1500 в сравнении с EFV на фоне стандартной базисной АРТ, состоящей из двух нуклеозидных/нуклеотидных ингибиторов обратной транскриптазы (НИОТ/НтИОТ), у ВИЧ-1-инфицированных пациентов, ранее не получавших лечения. Проведение исследования было одобрено Министерством здравоохранения Российской Федерации 21 апреля 2014 г. (разрешение № 219), а также локальными этическими комитетами исследовательских центров.
Исследование имеет бесшовный адаптивный дизайн и проводится в 2 этапа. На 1-м этапе основной целью является выбор оптимальной дозы VM-1500 (20 мг или 40 мг в сутки) на фоне стандартной базисной АРТ по показателю снижения вирусной нагрузки на 12-й неделе до < 400 копий/мл у 90 ВИЧ-1-инфицированных пациентов, ранее не получавших лечения. На 2-м этапе основная цель – оценить эффективность VM-1500 в выбранной оптимальной дозе в сравнении с EFV на фоне стандартной базисной АРТ по показателю снижения вирусной нагрузки до неопределяемого уровня (< 50 копий/мл) на 24-й неделе у 120 ВИЧ-1-инфицированных пациентов, ранее не получавших лечения.
В исследовании был сформулирован ряд дополнительных целей.
 Вирусологических – среднее снижение РНК ВИЧ в течение 48 недель исследуемой терапии; доля пациентов, достигших снижения вирусной нагрузки в 10 раз (1 log10 копий/мл) и более на 4-й неделе; доля пациентов, достигших уровня вирусной нагрузки < 400 копий/мл на 12-й неделе; доля пациентов, продолживших прием исследуемой терапии до 48-й недели.
Вирусологических – среднее снижение РНК ВИЧ в течение 48 недель исследуемой терапии; доля пациентов, достигших снижения вирусной нагрузки в 10 раз (1 log10 копий/мл) и более на 4-й неделе; доля пациентов, достигших уровня вирусной нагрузки < 400 копий/мл на 12-й неделе; доля пациентов, продолживших прием исследуемой терапии до 48-й недели.
Иммунологических – изменение абсолютного числа СD4+- и СD8+- лимфоцитов в течение 48 недель терапии.
Оценка резистентности ВИЧ – доля пациентов, у которых в течение 48 недель развилась резистентность ВИЧ-1 к исследуемой терапии.
Оценка безопасности – частота развития НЯ разной степени тяжести по данным субъективных жалоб, физикального осмотра, жизненных показателей, лабораторных исследований, ЭКГ при подборе оптимальной дозы VM-1500; частота развития НЯ «особого интереса», в том числе нарушений со стороны центральной нервной системы.
Исследование фармакокинетики VM-1500 – определение фармакокинетического профиля VM-1500 у выборочного числа пациентов.
В исследование было включено 90 пациентов с ВИЧ- 1-инфекцией, ранее не получавших АРТ, которые были рандомизированы (1:1:1) в 3 группы: 1-я группа – 30 больных, получавших VM-1500 в дозе 20 мг в сутки; 2-я – 30 больных, получавших VM-1500 в дозе 40 мг в сутки; 3-я (группа сравнения) – 30 больных, получавших EFV по 600 мг в сутки. Кроме того, все пациенты получали 2 НИОТ/НтНИОТ – TDF/FTC.
В исследование включали мужчин и женщин в возрасте 18 лет и старше с серологически подтвержденной (ИФА и иммунный блотинг) ВИЧ-1-инфекцией, стабильным клиническим течением ВИЧ-инфекции (клинические стадии 1 или 2 по классификации ВОЗ), которым по решению Исследователя показано начало АРТ (согласно Сводному руководству ВОЗ по использованию АРП для лечения и профилактики ВИЧ-инфекции от 2013 г.), с уровнем РНК ВИЧ-1 в плазме крови ≥ 5000 копий/мл и количеством СD4+-лимфоцитов > 200 клеток/мкл на скрининге. Перед проведением скрининга все пациенты получали информационный листок пациента и подписывали информированное согласие на участие в исследовании. Кроме того, было получено согласие пациентов на использование в течение всего исследования адекватных методов контрацепции (презерватив со спермицидом).
Исходные характеристики пациентов представлены в табл. 1. Существенных различий между группами пациентов в исходных показателях обнаружено не было. Среди вторичных заболеваний, имевших место в анамнезе, 3,7–6,9% пациентов отмечали пневмонии, 6,9–11,1% – опоясывающий лишай, 3,3–14,8% – эрозии шейки матки, 3,3–10,3% – анемию. Запланированная длительность участия пациентов в исследовании составляет 54 недели (2 недели – скрининг, 48 недель – исследуемая терапия и 4 недели – наблюдение). После этого пациенты, получавшие VM-1500, смогут продолжить лечение изучаемым препаратом в дополнительной открытой части исследования (до 96 недель). Набор пациентов в исследование начат в августе 2014 г. Окончание исследования ожидается в декабре 2016 г.
К настоящему моменту завершен 1-й этап исследования и проведен промежуточный (через 12 недель лечения) анализ эффективности и безопасности терапии. 12 недель лечения завершили 100% больных 1-й группы, 93,3% – 2-й и 80% – 3-й. Из-за развития НЯ выбыл 1 (3,3%) пациент 2-й группы и 2 (6,7%) пациента 3-й группы.
Абсолютное и относительное содержание CD4+- и CD8+-лимфоцитов (определяли методом проточной цитометрии) и уровень РНК ВИЧ (ПЦР) исследовали на скрининге через 4, 8 и 12 недель АРТ, параметры анализа периферической крови и биохимического анализа крови – до лечения, через 4, 8 и 12 недель терапии.
Для оценки эффективности терапии оценивали модифицированную популяцию пациентов, получивших лечение, и популяцию пациентов по протоколу. Модифицированная популяция пациентов, получивших лечение (MITT = modified intent-to-treat), соответствовала всем рандомизированным пациентам, которые получили, по крайней мере, одну дозу исследуемого препарата или препарата сравнения и у которых имелось хотя бы одно измерение вирусной нагрузки ВИЧ-1 после исходного. Популяция MITT являлась основной для анализа. Популяции по протоколу (ПП-анализ) соответствовали все пациенты популяции MITT, которые полностью завершили исследование (или его этап), имели оценки для первичного анализа эффективности и считались комплаентными (не имевшими каких-либо серьезных нарушений протокола в ходе исследования).
Для тестирования гипотезы неуступающей эффективности был вычислен 95% двусторонний ДИ для разности двух пропорций по основной конечной точке и границе сопоставимости δ = -15%. Отвержение нулевой гипотезы и принятие альтернативной докажет, что тестируемый препарат не уступает по эффективности стандартной терапии.
Описательную статистику по вирусной нагрузке представляли на обычной шкале (десятичной) и логарифмической (десятичный логарифм). Для сравнения групп терапии по изменению уровня вирусной нагрузки была построена смешанная линейная модель.
Изменение уровня РНК ВИЧ и количества CD4+-лимфоцитов описывали по временным точкам (визитам) исследования как непрерывные величины. Для каждой временнóй точки (исключая исходный уровень) вычисляли изменение параметра относительно исходного уровня и представляли в таблицах описательной статистикой. Внутригрупповые изменения параметра тестировали с помощью t-теста Стьюдента (для нормально распределенных данных) или знакового критерия Вилкоксона [Манна–Уитни] (для данных, не имеющих нормального распределения). В качестве теста на нормальность распределения использовали тест Шапиро–Вилка.
Регистрацию НЯ проводили с момента подписания пациентом формы информированного согласия и до 30 дней после последнего визита пациента в исследовательский центр или проведения последней процедуры, предусмотренной протоколом. Все НЯ, зарегистрированные в индивидуальной регистрационной карте пациента, были разделены на явления, возникшие в ходе исследования, и все остальные. Учитывали всех пациентов, которые получили, по крайней мере, одну дозу исследуемого препарата или препарата сравнения. Сравнение было проведено для НЯ «особого интереса», развившихся в ходе исследования. Согласно протоколу, к НЯ «особого интереса» были отнесены нарушения со стороны нервной системы (Nervous system disorders) и психиатрические нарушения (Psychiatric disorders). Группы терапии сравнивали по частоте встречаемости данных НЯ (рассматривали число пациентов с НЯ «особого интереса»). Данный параметр представляли в абсолютных и относительных значениях.
Исследование проведено при финансовой поддержке ООО «Вириом» (г. Химки, Московская область). Проведение исследования было организовано контрактно-исследовательской организацией ООО «ИФАРМА» (г. Химки, Московская область).
Результаты
Через 4 недели АРТ медиана РНК ВИЧ-1 была равна 2,5–2,6 log10 копий/мл (p < 0,001 для всех групп через 4 и 12 недель по сравнению с исходными значениями), через 12 недель – 1,7–1,8 log10 копий/мл (p < 0,001 для всех групп по сравнению с исходными значениями). Спустя 12 недель терапии доля больных с уровнем РНК ВИЧ-1 < 400 копий/ мл составила: MITT-анализ – 93,3, 86,2 и 81,5%; ПП-анализ – 93,1, 86,2 и 87,5% соответственно (рис. 1). По данному показателю VM-1500 в дозе 20 мг на 11,8%, а в дозе 40 мг – на 4,7% превосходил EFV (нижняя граница 95% ДИ -2,6 и -11,5% соответственно, что в обоих случаях правее δ = -15%). Таким образом, VM-1500 в дозах 20 и 40 мг в сутки достоверно не уступал по эффективности EFV по снижению вирусной нагрузки до уровня < 400 копий/мл за период 12 недель.
Дополнительный анализ снижения медианы содержания РНК ВИЧ через 12 недель терапии в зависимости от исходного уровня вирусной нагрузки (≥ 100 000 копий/мл и < 100 000 копий/мл) не выявил существенных различий между показателями у пациентов всех трех групп как при исходно высоком, так и при исходно низком уровне РНК ВИЧ. У больных с исходно низким уровнем РНК ВИЧ (медиана – 4,68–4,87 log10 копий/мл) спустя 12 недель терапии медиана показателя снизилась на 2,95–3,15 log10 копий/мл, а у больных с исходно высоким уровнем вирусной нагрузки (медиана – 5,38–5,63 log10 копий/мл) – на 3,28–3,38 log10 копий/мл.
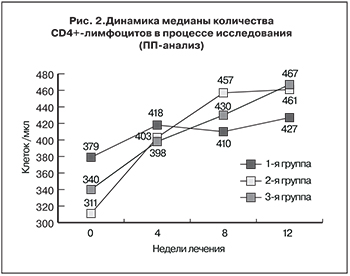
Динамика медианы количества CD4+-лимфоцитов представлена на рис. 2. Через 4 недели АРТ медиана количества CD4+-лимфоцитов варьировала от 398 до 418 клеток/мкл, а к 12 неделям лечения составила 427–467 клеток/мкл. Среднее количество CD4+-лимфоцитов через 12 недель терапии у больных 1-й группы увеличилось с 385,9 ± 174,7 до 486,2 ± 201,0 клеток/мкл (p < 0,001), 2-й группы – с 335,0 ± 97,4 до 459,1 ± 125,0 клеток/мкл (p < 0,001), 3-й группы – с 427,1 ± 225,5 до 504,6 ± 220,0 клеток/мкл (p = 0,037). Только у пациентов 1-й группы, получавших 20 мг VM-1500 в сутки, спустя 12 недель лечения отметили существенное уменьшение среднего количества CD8+-лимфоцитов (с 1076,2 ± 462,9 до 931,4 ± 370,4 клеток/ мкл; p < 0,001). У больных других групп изменения этого показателя были недостоверны. Вместе с тем у пациентов всех групп регистрировали увеличение медианы иммунорегуляторного индекса (соотношение CD4+/CD8+-лимфоцитов) с 0,357–0,378 до лечения до 0,487–0,522 после лечения.
Таким образом, предварительные результаты исследования свидетельствуют о сопоставимой (неуступающей) эффективности схем АРТ, включавших препарат VM-1500 в суточных дозах 20 и 40 мг, и схемы, содержавшей EFV. В то же время при проведении MITT-анализа через 12 недель терапии было показано, что в 1-й группе доля больных с уровнем РНК ВИЧ < 400 копий/мл была на 11,8% выше, чем в 3-й группе.
НЯ любой степени тяжести были зарегистрированы у 70,0, 86,2 и 85,7% больных соответственно. НЯ тяжелой степени были отмечены у 6,7, 13,8 и 14,3% пациентов, а НЯ, связанные с лечением, – у 3,3, 13,8 и 46,4% больных соответственно (табл. 2)
У 3 больных были установлены серьезные НЯ (СНЯ), маловероятно связанные с проводимой терапией. В 1-й группе у 1 больного после переохлаждения была выявлена пневмония нижней доли правого легкого легкой степени тяжести, еще у 1 – камень средней чашечки правой почки умеренной степени тяжести. В 3-й группе у 1 больного после переохлаждения была диагностирована пневмония верхней и средней доли правого легкого умеренной степени тяжести. Развитие СНЯ у всех трех больных не потребовало отмены или изменения схемы АРТ.
В данном исследовании к НЯ «особого интереса» были отнесены неврологические и психиатрические нарушения, которые имели место у 26,7% больных 1-й группы, у 44,8% – 2-й и 57,1% – 3-й (табл. 3). Нарушения со стороны ЦНС были выявлены у 20,0, 37,9 и 53,6% больных соответственно. При этом НЯ тяжелой степени не были отмечены у пациентов 1-й группы, тогда как во 2-й и 3-й группах их обнаруживали у 6,9 и 10,7% больных. Лишь у 1 (3,3%) больного 1-й группы и у 2 (6,9%) больных 2-й группы неврологические нарушения были связаны с исследуемым препаратом, в то время как в 3-й группе связь НЯ с приемом EFV была установлена у 12 (42,9%) больных. Среди неврологических НЯ, частота регистрации которых составляла 5% и более, у пациентов 1-й группы отметили только появление головной боли; у пациентов 2-й группы – необычные сновидения, головокружение, головную боль, нарушения сна, сонливость, а у пациентов 3-й группы (помимо указанных выше нарушений) – бессонницу и нарушения памяти.
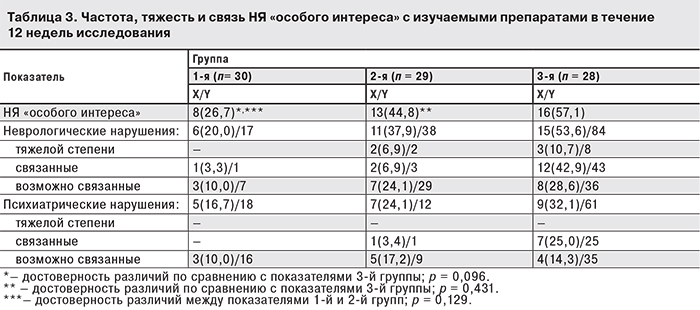
Психиатрические нарушения были обнаружены у 16,7, 24,1 и 32,1% больных соответственно. Ни у одного из пациентов не были выявлены НЯ тяжелой степени (см. табл. 3). Наиболее часто (≥ 5%) у больных 1-й группы наблюдали развитие депрессии, кошмаров, нарушений сна; у больных 2-й группы имели место только нарушения сна. Спектр психиатрических НЯ у больных 3-й группы включал агрессивность, апатию, депрессию, нарушения внимания, раздражительность, перепады настроения, кошмары, нарушения сна. Необходимо отметить, что у 25% больных 3-й группы эти НЯ были определенно связаны с приемом EFV, а еще у 14,3% – возможно связаны. Среди больных, получавших VM-1500, лишь у 1 (3,4%) пациента нарушения сна были связаны с приемом исследуемого препарата.
НЯ со стороны желудочно-кишечного тракта регистрировали в 1-й группе у 13,3% больных , во 2-й – у 41,4% и в 3-й – у 32,1%. Диарею легкой и средней тяжести отмечали у 17,2% пациентов 2-й группы, легкой степени тяжести – у 7,1% пациентов 3-й группы; тошноту легкой, средней и тяжелой степени – у 17,2% больных 2-й группы, легкой степени тяжести – у 7,1% больных 3-й группы. Во 2-й группе развитие НЯ было связано с приемом исследуемого препарата в 13,8% случаев.
Развитие НЯ со стороны кожных покровов наблюдали только у 2 (6,6%) пациентов 1-й группы (лишь в 1 случае имела место возможная связь с лечением). Среди пациентов 3-й группы подобные НЯ регистрировали в 42,9% случаев (в 32,2% отмечена связь с принимаемым препаратом).
Оценка изменений показателей периферической крови показала, что наиболее частыми НЯ в течение первых 12 недель лечения были лейкопения и нейтропения. Развитие лейкопении легкой степени тяжести регистрировали у 13,3% пациентов 1-й группы и у 7,1% пациентов 3-й группы, легкой и средней степени тяжести – у 13,8% пациентов 2-й группы. Наличие нейтропении легкой степени тяжести отмечали у 3,4–6,7% больных 1-й и 2-й групп и у 14,3% пациентов 3-й группы.
Поскольку в исследование не включали больных, страдающих вирусными гепатитами, то до начала исследования только у 3,6–6,7% пациентов были повышены уровни АлАТ и/или АсАТ 1-й степени токсичности (менее чем в 2,5 раза выше верхней границы нормы). В течение 12 недель исследования каких-либо существенных колебаний уровней АсАТ и АлАТ у больных всех групп выявлено не было. Другие показатели биохимического анализа крови (уровни креатинина, мочевины, альбумина, гамма-ГГТ, ЛДГ, КФК, холестерина, глюкозы) изменялись незначительно. Ни в одном случае не потребовалось коррекции параметров биохимического анализа крови.
Таким образим, схемы АРТ, включавшие VM-1500 в дозах 20 мг и 40 мг в сутки в сочетании с TDF/FTC, были так же эффективны, как и схема EFV + TDF + FTC, вне зависимости от исходного уровня вирусной нагрузки. Снижение уровня РНК ВИЧ в течение 12 недель терапии составило 2,9–3,2 lg копий/мл. Доля пациентов с уровнем РНК ВИЧ < 400 копий/мл была максимальной в 1-й группе и составила 93,3%, что на 11,8% выше, чем в группе сравнения (MITT-анализ). Доказана сопоставимая (неуступающая) эффективность обеих доз VM-1500 и EFV в схемах АРТ. При использовании всех терапевтических режимов отмечена иммунологическая эффективность лечения. Прирост количества CD4+-лимфоцитов (по медиане) у больных, получавших VM-1500, составил 48–150 клеток/ мкл, а у пациентов, получавших EFV, – 120 клеток/мкл. Увеличение количества CD4+-лимфоцитов обусловило повышение значений иммунорегуляторного индекса (соотношение CD4+/CD8+-лимфоцитов), в большей степени отмеченное у пациентов 1-й группы.
Безопасность применения в течение 12 недель схем АРТ, включавших VM-1500, была выше, чем при использовании схемы, включающей EFV. В 1-й группе частота всех НЯ средней (26,7%) и тяжелой степени (6,7%) была минимальной в сравнении с пациентами 3-й группы (60,7 и 14,3% соответственно). Развитие НЯ с проводимой терапией в 1-й группе было определенно связано только у 3,3% больных, возможно связано – у 23,3%, тогда как среди больных 3-й группы – у 46,4 и 50,0% соответственно.
Лишь у 3 пациентов были зарегистрированы 3 СНЯ, не связанные с приемом исследуемых препаратов и не потребовавшие отмены или изменения схемы лечения.
Развитие НЯ «особого интереса» (нарушения со стороны ЦНС и психической деятельности) у пациентов 1-й и 2-й групп выявляли существенно реже по сравнению с больными 3-й группы – в 26,7, 44,8 и 57,1% случаев соответственно. У пациентов 1-й группы также реже отмечали развитие НЯ со стороны желудочно-кишечного тракта. В течение первых 12 недель исследования не было выявлено существенных изменений показателей биохимического анализа крови и параметров периферической крови.
На основании проведенного анализа данных для дальнейшего исследования была выбрана доза препарата VM-1500 20 мг в сутки.


