Значительный прогресс в разработке и внедрении новых антиретровирусных препаратов (АРВП) позволил существенно изменить подход к лечению ВИЧ-инфекции. Обладая высокой эффективностью, новые режимы для начала терапии стали менее токсичны и более удобны для приема пациентами, чем режимы, использовавшиеся в начале эры антиретровирусной терапии (АРВТ). Новые препараты, представляющие следующие поколения известных классов, а также представители новых классов с более благоприятным профилем безопасности как в краткосрочной, так и в отдаленной перспективе позволили специалистам внедрять новые подходы к ведению пациентов, такие как переключение с более токсичного препарата предыдущего поколения на новый препарат этого же или другого класса с целью длительного сохранения эффективности АРВТ путем повышения приверженности к лечению.
Цель данного обзора – рассказать о новом представителе класса ННИОТ рилпивирине, который, возможно, найдет применение именно в качестве препарата переключения с режимов, содержащих представителей 1-го поколения ННИОТ [эфавиренз (EFV), невирапин (NVP)] и др.
Рилпивирин (RPV, TMC278, EDURANT, «Янссен Фармасьютикалс», Бельгия) был недавно зарегистрирован в США, Канаде и Европе в качестве АРВ-препарата 1-й линии, а также в качестве средства в виде «одной таблетки» в комбинации с тенофовира дизопроксила фумаратом (TDF) и эмтрицитабином (FTC) (комплера, эвиплера) [1]. В России препарат находится в процессе регистрации. Следует отметить, что любые новые АРВП должны обладать, по крайней мере, не худшей эффективностью и лучшей безопасностью. В связи с этим RPV широко изучали в сравнении с EFV, который представляет собой современный стандарт лечения [2]. В настоящее время в развитых странах комбинированный препарат атрипла (EFV/TDF/FTC) является наиболее часто назначаемым у пациентов, ранее не получавших лечения [3]. RPV также входит в состав комбинированного препарата эвиплера, который позиционируется как альтернатива атрипле у тех пациентов, кому не показано назначение EFV. RPV также позиционируется в качестве препарата переключения с EFV для уменьшения риска развития нежелательных явлений, поскольку профиль безопасности его несколько лучше, что продемонстрировали исследования, результаты которых представлены ниже.
Клинические исследования фазы II
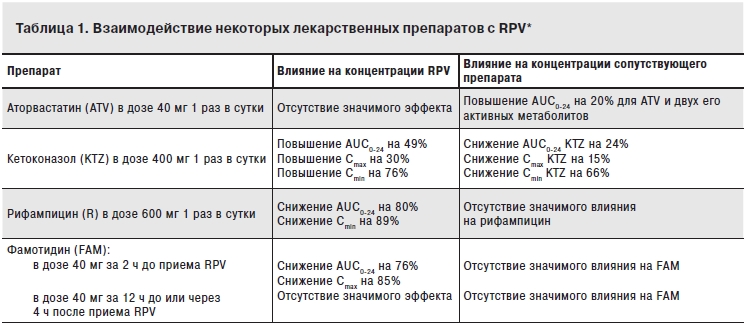
Фармакокинетику и вирусологическую активность RPV при приеме 1 раз в сутки изучали в пилотном рандомизированном двойном слепом исследовании фазы IIa TMC278-C201, целью которого было определение оптимальной дозы препарата [4]. В 5 групп терапии было рандомизировано 47 пациентов, которые принимали плацебо или RPV в дозах 25, 50, 100 или 150 мг в качестве монотерапии в течение 7 дней. В исследование были включены взрослые больные ВИЧ-1-инфекцией, не получавшие ранее АРВТ. Исходно РНК ВИЧ в плазме крови была >5000 копий/мл, а число CD4+-лимфоцитов – от 75 до 500 клеток/мкл. По демографическим данным и характеристикам заболевания группы были сопоставимы. В исследовании была продемонстрирована быстрая и существенная абсорбция RPV. Отмечалось достижение Сmax в течение 3–4 ч, а конечный период полувыведения составлял 48 ч, благодаря чему был возможен прием препарата 1 раз в сутки. Изменение вирусной нагрузки от исходных значений к 8-му дню было значительно больше во всех группах RPV по сравнению с плацебо (p<0,01). Вирусологический ответ был сходным в группах активной терапии RPV. Медиана снижения вирусной нагрузки к 8-му дню в группах терапии составила -1,199 log10 по сравнению с +0,002 в группе плацебо. Доля пациентов в группах терапии, у которых отмечалось уменьшение вирусной нагрузки более чем на 1 logl0, составила 78%, в группе плацебо таких изменений не отмечено. Частоту лекарственной резистентности и изменения числа CD4+-клеток нельзя было оценить достоверно с учетом небольшой продолжительности этого исследования. В процессе терапии не отмечено нежелательных явлений, которые бы приводили к перерыву в лечении или его полному прекращению. Подобно другим препаратам из группы ННИОТ, RPV является одновременно субстратом и индуктором CYP3A4 [1, 3]. В связи с этим данный препарат может вступать во взаимодействия с другими лекарственными препаратами (табл. 1). Так, отмечено снижение концентрации TDF примерно на 1/4, тогда как на фоне сопутствующего приема дарунавира/ритонавира – напротив, повышение концентраций RPV примерно в 2 раза [3]. Считается, что ни одно из этих взаимодействий не будет иметь значительных клинических последствий. При использовании RPV в сочетании с неантиретровирусными препаратами результаты взаимодействий сходны с EFV и NVP [3]. Отличительной особенностью RPV (как и атазанавира) является зависимость его всасывания в желудочно-кишечный тракт от кислотности желудочного сока. В связи с этим его одновременный или с интервалом не более 2 ч прием с ингибиторами H2-гистаминовых рецепторов (например, FAM) приводит к снижению сывороточной концентраций RPV на 75% [5]. Поэтому рекомендуется принимать FAM за 12 ч до или через 4 ч после приема RPV. Одновременный прием RPV с ингибиторами протонной помпы противопоказан. Важно также обязательно принимать RPV с пищей, так как прием пищи повышает секрецию соляной кислоты париетальными клетками, что усиливает всасывание препарата в желудочно-кишечный тракт.
Примечание. *Доза RPV – 150 мг 1 раз в сутки. AUC0-24 – площадь под кривой зависимости концентрации от времени (от 0 до 24 ч); Cmax – максимальная концентрация; Cmin – минимальная концентрация; AUC0-24 – площадь под кривой зависимости концентрации от времени (от 0 до бесконечности).
В исследовании фазы IIb TMC278-204 RPV вновь продемонстрировал мощную вирусологическую и иммунологическую активность в разных дозах в сравнении с EFV – текущим стандартом терапии ННИОТ [1]. Исследование было рандомизированным, частично слепым, активно контролируемым; в ходе его оценивали взаимосвязь между дозой и эффективностью, а также профилем безопасности RPV в течение 96 нед терапии [6]. В данном исследовании 368 ВИЧ-1-инфицированых пациентов старше 18 лет, ранее не получавших лечения, имевших исходно вирусную нагрузку > 5000 копий/мл, были рандомизированы в группы терапии RPV в дозах 25, 50 или 150 мг в сутки или EFV в дозе 600 мг в сутки. В качестве базовой терапии во всех группах использовали комбинации ламивудин/зидовудин или FTC/TDF. Первичной конечной точкой исследования была оценка эффективности терапии в виде количества пациентов, у которых вирусная нагрузка достигала <50 копий/мл или сохранялась на уровне <50 копий/мл после 96 нед терапии. К вторичным конечным точкам относились фармакокинетика, безопасность, иммунный ответ и развитие лекарственной резистентности. Демографические показатели и характеристики заболевания были сопоставимы в группах сравнения. Вместе с тем, доля пациентов с исходной вирусной нагрузкой >300 000 копий/мл была выше в группах пациентов, получавших RPV.
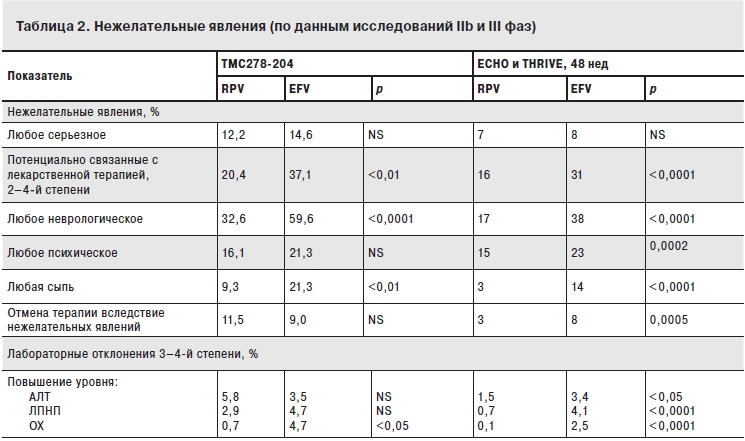
Вирусологическая эффективность была одинакова во всех исследуемых группах. Уровень РНК ВИЧ <50 копий/мл был достигнут в группах RPV в дозах: 25 мг – у 76% пациентов, 75 мг – у 72%, 150 мг – у 71% и у 71% в группе EFV). Доля пациентов с вирусологической неэффективностью также была сходной между группами (6% в группах RPV и 7% в группе EFV). Достоверных различий по иммунологической эффективности через 96 нед лечения также не было обнаружено: среднее повышение числа CD4+-клеток от исходных значений составило 159 клеток/мкл в группах RPV и 160 клеток/мкл в группе EFV. Эквивалентная эффективность RPV и EFV поддерживалась в течение 192 нед. Несмотря на сходную эффективность, наблюдались различия по безопасности исследуемых схем АРВТ. Так, в группе EFV чаще наблюдались побочные эффекты со стороны нервной системы, сыпь и повышение уровня холестерина. Нежелательные явления, приводившие к отмене терапии, встречались редко и с равной частотой в группах RPV и EFV – 11,5 и 9,0% соответственно (табл. 2). При сравнении результатов в группах с разными дозами RPV было выявлено, что случаи отмены терапии вследствие нежелательных явлений чаще встречались в группах терапии RPV в дозах 75 и 150 мг по сравнению с группой терапии в дозе 25 мг. Поэтому эта доза была выбрана для проведения дальнейших исследований в рамках фазы III.
Примечание. АЛТ – аланинаминотрансфераза, ЛПНП – липопротеиды низкой плотности, NS – незначимый, ОХ – общий холестерин.
Клинические исследования фазы III. Эффективность
В рамках фазы III были проведены 2 рандомизированных двойных слепых активно контролируемых многоцентровых исследования (ECHO и THRIVE) продолжительностью 96 нед по сравнению эффективности и безопасности RPV и EFV у ВИЧ-1-инфицированных пациентов, не получавших ранее АРВТ [7, 8]. Критерии включения и исключения были сходными с таковыми в исследовании фазы IIb. Дополнительным критерием исключения было наличие документированной информации по меньшей мере об одной из 39 мутаций, ассоциированных с резистентностью к ННИОТ (A98G, L100I, K101E/P/Q, K103H/N/S/T, V106A/M, V108I, E138A/G/K/Q/R, V179D/E, Y181C/I/V, Y188C/H/L, G190A/C/E/Q/S/T, P225H, F227C, M230I/L, P236L, K238N/T и Y318F), любое клинически значимое заболевание в активной форме (например, панкреатит, надпочечниковая недостаточность, заболевание сердца, нарушение функции печени, психическое заболевание), нарушение функции почек (установленная скорость клубочковой фильтрации по креатинину <50 мл/мин), а также беременность или грудное вскармливание.
В двух исследованиях 1368 пациентов были рандомизированы в соотношении 1:1 в группу терапии RPV в дозе 25 мг 1 раз в сутки и группу терапии EFV в дозе 600 мг в 1 раз в сутки. В исследовании ECHO в качестве нуклеозидной основы все пациенты получали TDF/FTC. В исследовании THRIVE нуклеозидную основу выбирал врач-исследователь, при этом 60% пациентов получали TDF/FTC, 30% – ламивудин/зидовудин, 10% – абакавир/ламивудин. Первичной конечной точкой исследования было достижение не меньшей эффективности RPV (по сравнению с EFV) в рамках подтвержденного вирусологического ответа на 48-й неделе (% пациентов, которые достигли вирусной нагрузки <50 копий/мл). Не меньшая эффективность определялась в том случае, если разница в эффективности двух препаратов на 48-й неделе не превышала порог в 12% при 95% доверительном интервале (ДИ). Избранный предел не меньшей эффективности был выбран в соответствии с рекомендациями Управления по контролю за пищевыми продуктами и лекарственными средствами США (FDA) для разработки АРВП, согласно которым предел должен составлять от 10 до 12%. Вторичные конечные точки исследования включали безопасность и переносимость лечения, генотипические и фенотипические характеристики ВИЧ (при вирусологической неэффективности), комплаентность (согласно оценке по модифицированному самоопроснику по приверженности терапии M-MASRI), фармакокинетику, а также взаимосвязь между фармакокинетикой и фармакодинамикой. Не ответившими на лечение считались пациенты, у которых терапия была преждевременно отменена по любой причине, либо отмечалась вирусологическая неэффективность терапии.
Исходные характеристики были сходными в рамках каждого исследования, а также при объединении исследований. Большинство (60–62%) больных были европеоидной расы. Средний возраст по медиане составил 36 лет. В исследовании принимали участие 20–28% женщин. Медиана количества CD4+-лимфоцитов составляла 256 клеток/мкл, а медиана исходной РНК ВИЧ – 5 log10 копий/мл. Уровень РНК ВИЧ > 100 000 копий/мл имели 45–53% больных. У 5–10% пациентов диагностировали вирусные гепатиты В и/или С. Пациенты были разделены по вирусной нагрузке при скрининге (≤100 000 копий/мл, >100 000 копий/мл, ≤500 000 копий/мл и >500000 копий/мл).
Поскольку дизайн исследований ECHO и THRIVE был схож, на 48-й неделе был сделан объединенный анализ (табл. 3). Было показано, что RPV не менее эффективен, чем EFV, при этом вирусная нагрузка <50 копий/мл была достигнута у 84,3% пациентов в группе RPV и у 82,3% – в группе EFV. Кроме того, в исследуемых группах наблюдали одинаково стабильное увеличение числа CD4+-клеток по сравнению с исходными значениями. Общие показатели частоты неэффективности терапии были равными в группах терапии, тогда как причины неэффективности различались. Неэффективность лечения в группах RPV ассоциировалась с недостаточным подавлением репликации РНК ВИЧ, в то время как среди пациентов в группах EFV чаще отмечалась неэффективность вследствие токсичности. Последующий анализ показал, что вирусологическая неэффективность чаще встречалась среди пациентов, получавших RPV, с недостаточной приверженностью. Так, в исследовании ECHO среди пациентов с приверженностью ≤95% доля ответивших на лечение в группах RPV (30 из 44) и EFV (41 из 56) составила соответственно 68 и 73%. Доля больных с исходной вирусной нагрузкой ≤100 000 копий/ мл, ответивших на лечение, была выше в группах RPV и составила 90% в исследовании ECHO и 91% в исследовании THRIVE (в группах EFV соответственно 83 и 84%). Доля пациентов с исходной РНК ВИЧ от 100 000 до 500 000 копий/мл, ответивших на лечение, в группах RPV составила 79% (ECHO) и 80% (THRIVE), в группах EFV – 83% (ECHO) и 82% (THRIVE). Тот же показатель среди больных с исходно высокой вирусной нагрузкой (>500000 копий/мл) в исследовании ECHO был существенно ниже для RPV, чем для EFV (62 и 81% соответственно). В исследовании THRIVE, напротив, RPV превосходил EFV по вирусологической эффективности (77 и 69%, соответственно). Учитывая небольшую численность пациентов в группах, полученные данные необходимо интерпретировать с осторожностью. В настоящее время продолжается анализ результатов исследований для более полного понимания роли таких факторов, как приверженность лечению, экспозиция препарата и исходная вирусная нагрузка в развитии вирусологической неэффективности.
Примечание. NS – незначимый, NR – не описано.
Объединенный 96-недельный анализ ECHO и THRIVE подтвердил результаты 48-недельного первичного анализа, показавшего, что RPV не менее эффективен, чем EFV [8]. Анализ подгрупп, выделенных по полу, региону, этнической принадлежности, а также наличию гепатитов В и С, продемонстрировал сходную эффективность RPV и EFV, что свидетельствует о возможности более широкого применения этих данных.
Клинические исследования фазы III. Безопасность
Данные по безопасности и переносимости RPV получены преимущественно в исследованиях фаз IIb и III [2, 9]. В исследовании фазы IIb наиболее частыми нежелательными явлениями, которые связывали с применением препарата, были тошнота и головокружение (3,6% случаев для RPV и 1,1% – для EFV). Большинство наблюдавшихся побочных эффектов не зависели от дозы препарата. Однако нежелательные явления, приводившие к отмене терапии, чаще проявлялись при применении более высоких доз RPV (в основном это касалось повышения уровней аминотрансфераз). На фоне приема более высоких доз RPV также повышалась частота появления сыпи и увеличивался интервал QT (всех степеней), но это ни в одном случае не привело к отмене АРВТ. Сравнительный анализ безопасности RPV и EFV, по данным исследования фаз IIb и III, представлен в табл. 2.

Данные объединенных исследований ECHO и THRIVE показали, что применение RPV сопровождалось меньшей частотой нежелательных явлений по сравнению с EFV. Суммарную токсичность 2–4-й степени к 96-й неделе АРВТ зафиксировали у 16,9% больных, получавших RPV (33,1% в группах EFV; p<0,0001) [10]. В группах пациентов, получавших RPV, реже отмечались неврологические и психиатрические нежелательные явления, а также сыпь. Большинство неврологических и психиатрических симптомов были 1-й или 2-й степени тяжести, их распространенность снижалась через 4–8 нед терапии в обеих группах. При приеме RPV исследователи отмечали развитие сыпи, связанной с исследуемым препаратом, лишь у 4% пациентов, при приеме EFV – у 15% (p<0,0001). При этом в большинстве случаев наблюдали 1-ю или 2-ю степень токсичности. У пациентов, которым была продолжена терапия, сыпь регрессировала в обеих группах, за исключением 5 больных из группы EFV, которым лечение было отменено [8].
Согласно полученным результатам, RPV показал лучший липидный профиль и меньшую гепатотоксичность, чем EFV. Отклонения биохимических показателей 2–4-й степени наблюдали у 6% и 5–18% пациентов в группах RPV и EFV соответственно. В основном это касалось повышения уровней аминотрансфераз, общего холестерина, ЛПНП и триглицеридов, причем изменение последнего показателя встречалось только среди больных, принимавших EFV [5]. Вместе с тем, между группами не отмечалось различий по изменениям индекса атерогенности (соотношения общего холестерина и ЛПВП) к 48-й неделе [7]. Лабораторные отклонения 3-й и 4-й степени по уровню гемоглобина чаще встречались у больных, принимавших RPV, хотя отмечено восстановление показателей до исходных значений на 96-й неделе терапии. В группе RPV немного повышалась средняя концентрация сывороточного креатинина, тем не менее, концентрация оставалась стабильной в течение всего исследования (5,69–9,07 мкмоль/л), тогда как в группе EFV этот показатель практически не изменялся (0,10–2,38 мкмоль/л). Таким образом, в группе RPV скорость клубочковой фильтрации оставалась немного ниже исходной, но в пределах нормы (среднее снижение 8–11 мл/мин на 1,73 м2). Отклонений уровня креатинина 3-й или 4-й степени не регистрировали ни в одной из исследуемых групп. Кроме того, не было отмечено случаев отмены терапии вследствие нежелательных явлений со стороны почек. При использовании цистатина C (альтернативного индикатора функции почек) выявлено, что RPV не оказывает на почки клинически значимого влияния [7].
Отмена терапии, связанная с развитием нежелательных явлений, была зарегистрирована к 96-й неделе лечения у 4,1% пациентов, получавших RPV и у 8,5% пациентов, получавших EFV.
В целом, согласно имеющимся данным, RPV является безопасным и хорошо переносимым препаратом [2, 9]. Кроме того, при прямом сравнении с EFV было показано, что RPV реже вызывает побочные эффекты со стороны нервной системы, сыпь и липидные нарушения. Это улучшение профиля токсичности обеспечивает снижение частоты отмены терапии в клинических исследованиях и отражает одно из главных преимуществ применения RPV в качестве нового подхода к АРВТ.
Клинические исследования фаз II и III. Резистентность
Отличительной особенностью RPV является его значительная активность по отношению к вирусам, резистентным к ННИОТ 1-го поколения. RPV сохраняет мощную антивирусную активность при наличии таких мутаций, как L100I, K103N, Y181C и Y181L, а также при сочетании мутаций K103N + Y181C и L100I + K103N [11]. Так, в одном из исследований in vitro оценивалась частота вирусного прорыва на фоне терапии RPV, EFV и NVP [12]. При использовании EFV и NVP в концентрациях 1 мкмоль отмечался значительный вирусный прорыв в течение 2 недель, тогда как при применении RPV в течение 30 дней при концентрациях ≥ 40 нмоль развития резистентности не отмечено. Кроме того, мутаций устойчивости не было выявлено вплоть до снижения концентрации RPV до 10 нмоль. Несмотря на эти обнадеживающие данные in vitro, пока ограничен объем существующих клинических данных, которые бы позволили описать активность RPV и его устойчивость к развитию вирусной резистентности.
В исследовании фазы IIb у 17 (6%) пациентов, получавших лечение RPV, отмечена вирусологическая неэффективность терапии. У 9 (53%) из них на фоне лечения развились мутации, ассоциированные с резистентностью к ННИОТ (L100I, K101E, K103N, V108I, E138K, E138R, Y181C и M230L). Наиболее частой мутацией, связанной с резистентностью к терапии, была E138K. В группе больных, получавших EFV с вирусологической неэффективностью (7%), у 50% пациентов развились мутации резистентности к ННИОТ, среди которых самой частой была K103N [9].
В исследованиях фазы III ECHO и THRIVE на 48-й неделе 10% пациентов в группе RPV и 6% в группе EFV имели вирусологическую неудачу (ВН) [6]. У пациентов с исходной РНК ВИЧ < 100000 копий/мл частота ВН была одинаковой (по 5%) в группах RPV и EFV. Однако у пациентов с исходной вирусной нагрузкой >100 000 копий/мл частота ВН была достоверно выше (17%) в группе RPV, чем в группе EFV (7%; p< 0,0001). Анализ показал, что ВН на фоне АРВТ, содержащей RPV, была связана с конкретными параметрами генотипической и фенотипической устойчивости. Данные по резистентности были доступны к концу 48-й недели для 62 из 72 пациентов с неэффективностью терапии на основе RPV и у 28 из 39 пациентов с неэффективностью лечения EFV. Среди них у 63% пациентов, получавших RPV, и у 54% пациентов, получавших EFV, на фоне терапии возникло более 1 мутации резистентности к ННИОТ (различия не были статистически значимыми). Вместе с тем, у 68% пациентов, получавших RPV, и 32% пациентов, получавших EFV, развилось более 1 мутации резистентности к НИОТ (различия достоверны) [2, 13]. Наиболее частыми мутациями, ассоциированными с резистентностью, были E138K и M184I в группе RPV и K103N и M184V в группе EFV. Объединенный анализ показал, что в обеих лечебных группах мутации к НИОТ возникали чаще в случае исходно высокой вирусной нагрузки (> 100 000 копий/мл), но в группах RPV этих мутаций было больше. Зависимость частоты развития тех или иных мутаций от подтипа ВИЧ-1 (подтип В или не-В) не выявлено [6].
У 50% больных с вирусологической неэффективностью на фоне терапии RPV и развившейся резистентностью к данному препарату была отмечена фенотипическая резистентность к RPV (в сравнении с 43% фенотипической резистентности к EFV). У пациентов обеих групп развилась мутация M184V/I, которая определяла снижение чувствительности к эмтрицитабину и ламивудину, хотя данная мутация отмечалась чаще у пациентов, получавших RPV. Важной особенностью является то, что у больных с исходно высоким уровнем РНК ВИЧ, ВН при использовании RPV и наличием мутаций резистентности к RPV отмечена перекрестная устойчивость к EFV (у 90% пациентов), ETR (у 93%) и NVP (у 48%), тогда как при тех же условиях и использовании EFV отмечалась перекрестная устойчивость только к NVP (в 100% случаев) [6].
Поскольку в большинстве исследований RPV применяли у пациентов, ранее не получавших лечения, объем данных об активности RPV у больных с лекарственно-резистентными вирусами весьма ограничен. В небольшом пилотном исследовании изучали активность RPV у пациентов с неэффективностью предшествующей терапии и с ≥1 мутацией, ассоциированной с резистентностью к ННИОТ [14]. В исследовании анализировали результаты применения трех доз RPV (25, 50 и 150 мг) у 36 пациентов в течение 7 дней терапии. На 8-й день снижение вирусной нагрузки в плазме от исходных значений составило 0,84 log10 копий/мл (95% ДИ: -2,3, 0,4; p<0,001). Достоверных различий по вирусологической активности разных доз препарата не отмечали. Однако в связи с небольшой продолжительностью исследования выводы об антивирусной активности RPV у пациентов с мутациями резистентности к ННИОТ сделать было невозможно.
Перспективы применения RPV
Необходимо проведение дальнейших исследований для изучения причин вирусологической неэффективности RPV и возникновения отдельных мутаций при исходно высокой вирусной нагрузке. Кроме того, существует потребность оценки применения RPV у больных с опытом лечения, так как предрегистрационный анализ фазы III осуществлялся только среди пациентов, которым лечение ранее не проводили.
Большие перспективы имеет лекарственная форма, в которой 3 АРВП содержатся в одной таблетке для приема 1 раз в сутки (RPV/TDF/FTC, эвиплера). В настоящее время продолжается 48-недельное исследование 50 ВИЧ-инфицированных пациентов по переключению со стабильной терапии атриплой на лечение эвиплерой. Результаты 12 нед терапии свидетельствуют о том, что вирусологическая эффективность на фоне применения исследуемого препарата сохраняется у всех пациентов, при этом снижается частота побочных эффектов [15].
Проводятся исследования альтернативных лекарственных форм и путей введения RPV. Оценивали фармакокинетику RPV в виде гранул, которые позволяют легче вводить препарат и применять более гибкие дозы для детей. Согласно имеющимся данным, эта лекарственная форма обладает более приятным вкусом и сходной биодоступностью по сравнению с таблетками для взрослых [13]. RPV также изучают в качестве инъекционной лекарственной формы депо-препарата с замедленным высвобождением [16, 17]. Недостаточная приверженность является одной из частых причин неэффективности терапии. При использовании RPV в виде однократной внутримышечной или подкожной инъекции наносуспензии у 51 здорового добровольца наблюдали хорошую переносимость и обеспечение лекарственной экспозиции в течение нескольких месяцев [17]. На основании полученных многообещающих результатов запланированы дальнейшие исследования этой лекарственной формы.
Таким образом, класс ННИОТ остается важным элементом в терапии ВИЧ-инфекции, поэтому продолжаются разработки новых препаратов, которые по эффективности не уступают препаратам 1-го поколения, но имеют более благоприятный профиль безопасности. Один из таких препаратов, RPV, в двух исследованиях фазы III продемонстрировал не меньшую эффективность по сравнению с EFV. При этом частота побочных эффектов при приеме RPV была статистически достоверно ниже. Однако отмечено, что у пациентов с исходно высокой вирусной нагрузкой RPV несколько менее эффективен, чем у пациентов с исходной вирусной нагрузкой <100 000 копий/мл. Также была показана несколько бoльшая по сравнению с EFV зависимость эффективности RPV от степени приверженности, а также бoльшая частота развития мутаций устойчивости к ННИОТ и НИОТ на фоне неудачи лечения RPV. С учетом полученных данных можно предположить, что RPV в первой линии АРВТ может быть препаратом выбора для пациентов с исходной вирусной нагрузкой <100 000 копий/ мл, высокой приверженностью, возможностью принимать препарат с пищей и с особенностями, делающими нежелательным применение EFV. Необходимо продолжить изучение эффективности и безопасности режимов терапии с RPV, применяемых для переключения с EFV у пациентов с неопределяемой вирусной нагрузкой. Предварительные данные свидетельствуют о том, что эффективность терапии после переключения сохраняется у всех пациентов, при этом улучшается переносимость лечения, что позволяет надеяться на длительную эффективность АРВТ с сохранением в резерве большого арсенала других препаратов.

{kind=link}
{kind=link}
{kind=link}