Филогенетика, понимаемая как изучение эволюционной истории видов и организмов, генов и белков, в последние годы практически превратилась в отдельное направление биологии. Применяемая к изучению эволюции и распространения патогенных микроорганизмов (вирусов, бактерий и т. д.), она становится также важной частью эпидемиологии. Базовым методом этого направления является установление филогенетических связей между исследуемыми объектами на основе типичных для них нуклеотидных или аминокислотных последовательностей. Такие связи визуализируются чаще всего в виде филогенетических деревьев [1, 2]. Применявшиеся методы можно, с известной долей условности, разделить на 2 группы: алгоритмические и оптимизирующие. Первые используют заранее сформулированную цепочку правил (алгоритм) для построения финального филогенетического дерева, которое, впрочем, может не соответствовать наиболее вероятному ходу реконструируемой эволюции. Из этих методов наиболее известны и популярны neighbour-joining (NJ) и unweighted pair group method using arithmetic averages (UPGMA). Вторые ищут, часто путем прямого перебора всех вариантов, дерево, оптимальное с точки зрения некоторой целевой функции. Метод максимальной парсимонии минимизирует число шагов эволюции (замен в геноме). Метод максимального правдоподобия подбирает изначально неизвестные параметры эволюционной модели так, чтобы максимизировать соответствующую функцию правдоподобия. Алгоритмические методы, как правило, используют понятие численного «генетического расстояния» (между нуклеотидными или аминокислотными последовательностями). К настоящему времени предложены десятки способов расчета таких расстояний, предполагающие различные модели мутационного процесса – от простейших до многопараметрических. Оптимизирующие методы, как правило, рассматривают нуклеотидные или аминокислотные последовательности как «текстовые» последовательности символов [1–4].
В последние годы наиболее адекватными методами филогенетики считаются методы в рамках Байесовского подхода (Bayesian inference), в частности, «расчеты методом Монте-Карло по схеме Марковской цепи» (Bayesian Markov chain Monte Carlo approach – MCMC). MCMC сочетает в себе элементы как алгоритмических методов, используемых для построения марковских цепей эволюционных событий, так и оптимизирующих методов, максимизируя так называемые апострериорные вероятности используемых параметров модели и моделируемых событий. В целом преимуществом Байесовских подходов является то, что параметры модели рассматриваются как переменные, имеющие статистическое распределение, а не как константы в методе максимального правдоподобия. При анализе этих распределений естественным образом возникают оценки достоверности параметров и результатов моделирования, например, оценки доверительного интервала времени происхождения того или иного эволюционного события (расположения узла на филогенетическом дереве) [2–5].
На современном этапе филогенетический анализ проводится с помощью специализированных, чаще всего общедоступных, компьютерных программ, наиболее популярные из которых перечислены в работах В.В. Лукашова [1] и Z. Yang и соавт. [4]. Впрочем, ежегодно появляются новые программы или улучшенные варианты уже известных [5, 6]. Каждый из методов филогенетического анализа обладает своими особенностями, достоинствами и недостатками, характерными задачами и областями применения и т. п., которые мы не имеем возможности обсуждать здесь детально, отсылая читателя к специальной литературе [1–12].
Как самостоятельный подраздел филогенетики патогенных микроорганизмов с 2004 г. развивается филодинамика, учитывающая влияние на эволюцию патогенов их механизмов передачи, особенностей взаимодействия с иммунной системой хозяев, скорости роста (или уменьшения) популяции патогена и других ключевых элементов эпидемического процесса [7, 8]. Отчасти филодинамика пересекается с филогеографией, основной задачей которой является выяснение места происхождения того или иного организма или вида (в том числе патогенных) и путей его последующей миграции и внедрения на новые территории [9, 10]. Своевременное выявление происхождения того или иного штамма особенно важно для тех видов патогенов, которые могут легко и быстро переноситься по миру мигрирующими птицами или в результате перемещений людей и домашних животных.
Для теоретической и практической эпидемиологии существенными оказались многие результаты филогенетических исследований вирусов: описание эпидемиологии и распространения ВИЧ; выяснение происхождения и последующей эволюции коронавируса SCoV, вызывающего ТОРС; отслеживание миграции вирусов птичьего гриппа, а также сезонных и пандемических штаммов вируса гриппа человека; уточнение путей заноса различных штаммов вируса лихорадки Западного Нила в Евразию и Северную Америку и т. п. [1, 3, 11, 12].
В Российской Федерации исследования в области филогенетики вирусов также ведутся достаточно широко. В частности, сотрудниками Центрального НИИ эпидемиологии Роспотребнадзора опубликованы работы по филогенетике и филогеографии вирусов птичьего гриппа и гриппа человека, хантавирусов, вирусов Крымской-Конго геморрагической лихорадки, клещевого энцефалита, лихорадки Западного Нила и др. [13–20]. Однако следует отметить, что, как правило, в работах российских ученых для выявления филогенетических связей вирусных штаммов используются простейшие методы типа NJ и UPGMA, а анализ их происхождения и эволюции сводится к качественным рассуждениям.
В данной публикации мы рассмотрим методологию и результаты проведенного нами совместно с итальянскими коллегами анализа эволюционной истории вируса Омской геморрагической лихорадки (ОГЛ). Этот пример удачен тем, что данная инфекция приобрела эпидемическую значимость и была идентифицирована уже в нашу эпоху, фактически на глазах современных ученых. При этом время и место, в которые вирус приобрел патогенность для человека, оставались не выясненными. Решение таких проблем представляет собой классическую задачу филогенетики и филогеографии, которая успешно может решаться методами типа MCMC.
ОГЛ – тяжелая вирусная зоонозная природно-очаговая болезнь, впервые выявленная в 40-х годах прошлого века в Сибири. Возбудитель ОГЛ – вирус ОГЛ (ВОГЛ), принадлежит к роду Flavivirus семейства Flaviviridae. Кроме ВОГЛ, геморрагические лихорадки способны вызывать также такие флавивирусы, как вирус лихорадки леса Кьясанур и его геновариант вирус Алхурма. Главный механизм передачи ВОГЛ человеку – контакт с тканями и кровью инфицированных ондатр Ondatra zibethicus. Возможно заражение в результате присасывания клещей, а также водным путем через контаминированную воду [21, 22]. По-прежнему остается дискуссионным вопрос о том, какие виды позвоночных (предположительно – водяная полевка) являются основными хозяевами ВОГЛ и какие виды членистоногих (предположительно – клещи рода Dermacentor) могут быть ведущими переносчиками ВОГЛ в природных условиях. Однако известно, что в периоды эпидемической и эпизоотической активности природных очагов ОГЛ вирус изолировали, кроме ондатр и больных людей, от достаточно широкого круга членистоногих (иксодовые и гамазовые клещи, кровососущие комары) и позвоночных (полевки родов Microtus и Myodes и др.).
В период с 1946 по 1958 г. более 1000 клинических случаев ОГЛ было диагностировано в Омской, Новосибирской, Курганской и Тюменской областях, пик заболеваемости (около 600 случаев) пришелся на 1946 г. В период между 1972 и 1988 г. официальная регистрация заболеваемости ОГЛ отсутствовала («межэпидемический период»). Вновь активность природных очагов ОГЛ наблюдается с 1988 г., в 1989–2001 гг. было зарегистрировано 165 клинических случаев ОГЛ, наибольшее число диагностировано в 1990 и 1991 гг. – 29 и 41 лабораторно подтвержденных случаев соответственно. Эпидемическую вспышку ОГЛ в конце 40-х годов связывают с тем, что более 4000 особей североамериканских ондатр было выпущено на волю в этом регионе в 1935–1939 гг. с целью их дальнейшего использования для промысловой охоты. Вид оказался высокочувствительным к ВОГЛ, что привело к многочисленным эпизоотиям.
Филогенетическое происхождение и эволюционная история ВОГЛ не были изучены, поскольку в распоряжении специалистов находились единичные штаммы ВОГЛ [23]. Западные исследователи высказывают предположение, что ВОГЛ генетически наиболее близок к вирусу клещевого энцефалита (ВКЭ) и более удален от других видов флавивирусов, переносимых клещами [23, 24]. К этому же выводу мы пришли еще в начале 2000- х годов на основе изучения 14 штаммов ВОГЛ из коллекции Омского НИИ природноочаговых инфекций [25, 26].
С точки зрения молекулярной эпидемиологии ключевым вопросом является происхождение ВОГЛ: сформировался ли современный генотип ВОГЛ, патогенный для человека, в результате мутации или ряда мутаций незадолго до начала его эпидемического распространения (по аналогии с коронавирусом SCoV [3]) или этот вирус долгое время циркулировал в Сибири и приобрел эпидемическое значение в результате изменения экологических условий? В нашей работе для ответа на этот вопрос использованы результаты расширенного генотипирования 25 штаммов ВОГЛ и современные компьютерные методы филогеографической реконструкции.
Материалы и методы
Характеристика штаммов
В работе использованы данные о нуклеотидных последовательностях гена Е гликопротеина оболочки 25 штаммов ВОГЛ, выделенных с 1947 по 2007 г. в Омской, Новосибирской и Курганской областях. 18 штаммов были изолированы от ондатр, 2 – от больных людей, и по одному от клещей видов Dermacentor marginatus и Androlaelaps casalis, комаров Aedes subdiversus, красной полевки Myodes rutilus и полевки-экономки Microtus oeconomus. Экстракцию и реверсию вирусной РНК, последующую амплификацию кДНК осуществляли стандартными методами с использованием праймеров и реагентов производства Центрального НИИ эпидемиологии. Секвенирование участков генома ВОГЛ по двум цепям выполнено на секвенаторе ABIprism sequencer согласно инструкциям производителя (Applied Biosystems, СШA). Для 16 образцов были получены полные последовательности гена Е (1488 нт), а для 9 – частичные последовательности длиной 632 нуклеотида. Нуклеотидные последовательности размещены в базе GenBank с номерами доступа AF482341 – AF482354 и JX315605 – JX315615.
Методы компьютерной реконструкции филогенеза
Для выбора эволюционной модели нуклеотидных замен, наиболее удачно описывающей имеющийся набор данных, применяли программу ModelTest v. 3.6 [27]. Возможность правдоподобной реконструкции филогенетических событий по полученным нуклеотидным последовательностям была изучена и подтверждена с помощью программы TreePuzzle [28].
Построение датированных филогенетических деревьев и оценки скорости эволюции производили в рамках Байесовского подхода с помощью «расчетов методом Монте-Карло по схеме Марковской цепи» (Bayesian Markov chain Monte Carlo approach – MCMC), используя программу Beast v. 1.6.1 [29].
Сопоставление с использованием фактора Байеса (Bayes factor – BF), являющегося отношением величин «маргинального правдоподобия» двух сравниваемых моделей, показало, что предположение о некоррелированных, экспоненциально распределенных, «ослабленных часах» (relaxed clock), позволяющих скорости эволюции различаться в разных ветвях эволюционного дерева, лучше описывает данные, чем предположение о «точных часах» (strict clock) эволюции, при котором скорость эволюции является фиксированной величиной для всего дерева. С учетом требований филодинамики были рассмотрены 4 параметрические модели роста популяции ВОГЛ (отсутствие роста, экспоненциальный, логистический и экспансивный рост) и непараметрическая кусочно-постоянная интерполяция роста по модели «Байесовского горизонта» (Bayesian skyline plot – BSP). При использовании «ослабленных часов» наиболее адекватной была BSP-модель.
Эволюционные цепи состояли не менее чем из 15 x 107 шагов, то есть «поколений» ВОГЛ, проверяемых через каждые 15 x 103 шагов. Отбор наиболее качественных моделей эволюции проводили в программе Beast методом маргинального правдоподобия, опять же используя фактор Байеса как меру достоверности: использовали только модели, для которых 2lnBF > 6, то есть «убедительно или очень убедительно подтвержденные» по критерию, сформулированному R. Kass и соавт. [30]. Отобранные эволюционные деревья суммировали с помощью подпрограммы Tree Annotator программы Beast в дерево с «максимальной надежностью кладов» (maximum clade credibility), то есть в дерево, характеризующееся максимальными апостериорными вероятностями параметров. При этом для каждого клада и кластера оценивалось время существования «последнего общего предка» (most recent common ancestor – MRCA). Диапазон неопределенности датирующих оценок указывался в виде интервала, в который укладывается 95% максимальной плотности апостериорного распределения (95 % highest posterior density interval – 95% HPD ).
Для выявления мест происхождения и направления распространения штаммов эволюцию представляли в виде Марковского процесса, в котором время описывалось непрерывной переменной, а географические точки – дискретной. Для анализа набора нуклеотидных последовательностей гена Е ВОГЛ использовали модель «выбора переменных путем Байесовского стохастического поиска» (Bayesian Stochastic Search Variable Selection model) из программы BEAST [29].
Результаты и обсуждение
Оценки скорости эволюции и реконструкция филогенеза во времени
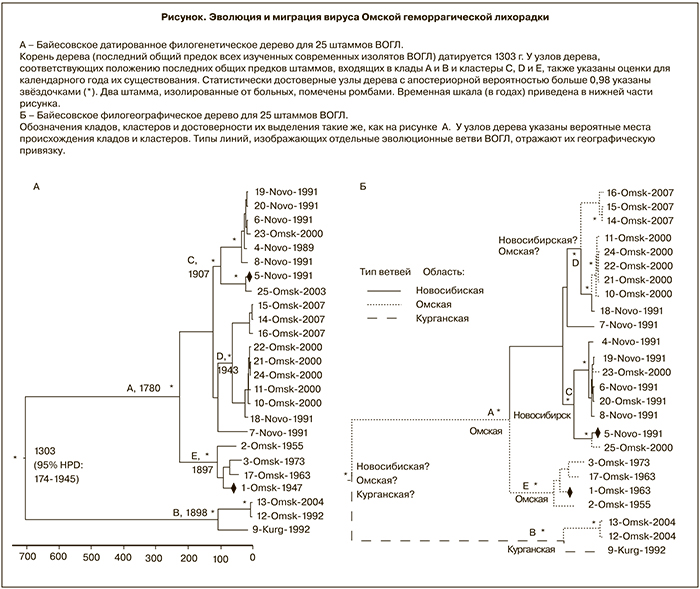
Средняя скорость эволюции гена Е ВОГЛ была оценена в 1,38 х 10-4 замен/на позицию/в год с 95% HPD от 1,36 х 10-5 до 3,03 х 10-4. На рисунке (А) показано датированное филогенетическое дерево с максимальной Байесовской надежностью кладов. Согласно вычислениям, последний общий предок для всех изученных штаммов ВОГЛ существовал 704 года назад, то есть в 1303 г. (95% HPD: 174–1945). В дальнейшем эволюционное дерево расщепляется на 2 основных клада, включающих по нескольку кластеров. Апостериорная вероятность такой классификации равна 0,99, то есть высоко статистически значима.
Общий предок клада А существовал в 1780 г. (95% HPD: 1466–1936). Внутри клада А статистически достоверно можно выделить 3 кластера (C, D, E). Кластер C, происхождение которого датируется 1907 г. (95% HPD: 1770–1985), включает 6 изолятов из Новосибирской области и 2 – из Омской. Кластер D, выделившийся в 1943 г. (95% HPD: 1857–1989), включает 8 изолятов из Омской области и 1 – из Новосибирской. Возникновение кластера E, в который входят 4 изолята из Омской области, датируется 1897 г. (95% HPD: 1808–1944). Отдельный клад B образован потомками общего предка, существование которого отнесено к 1898 г. (95% HPD: 1703–1991).
Филогеографический анализ
Филогеографическая интерпретация имеющихся данных не позволяет достоверно определить местонахождение общего предка современных штаммов ВОГЛ, поскольку помещает его в Омскую, Новосибирскую или Курганскую области практически с одинаковой вероятностью события (ВС): 0,37, 0,33 и 0,30 соответственно. Согласно расчетам, общий предок клада А находился на территории Омской области (ВС = 0,53), а клада B – в Курганской области (ВС = 0,47). Штаммы кластера C происходят из Новосибирской области (ВС = 0,63), а кластера E – из Омской области (ВС = 0,70). По имеющимся данным, происхождение кластера D может быть с равной вероятностью отнесено к Омской (ВС = 0,44) или Новосибирской области (ВС = 0,49). Анализ указывает и на вероятность миграции штаммов ВОГЛ между областями: так, например, 2 штамма «новосибирского» типа, включенные в кластер С, реально были выделены на территории Омской области (см. рисунок, Б).
Итак, первое выявленное нами эволюционное событие в истории ВОГЛ – расхождение кладов A и B – произошло около 700 лет назад. Кластеры C, D и E разошлись не ранее 1780 г. и продолжали эволюционировать в ХIХ и ХХ веках. Аналогичный анализ показал, что и вирусы лихорадки леса Кьясанур и Алхурма имели общего предка также 700 лет назад [31]. Для сравнения, общий предок для трех генотипов ВКЭ (европейского, сибирского и дальневосточного) существовал предположительно более 3000 лет назад [32, 33].
Маловероятно, что ВОГЛ эволюционировал где-то вне южных областей Сибири и был занесен туда вместе с ондатрами. Вирусы, подобные ВОГЛ, не обнаружены ни в Северной Америке (исходном месте обитания ондатр), ни где-либо еще, кроме Сибири.
Если бы ВОГЛ, циркулируя ранее исключительно среди сибирских резервуарных хозяев, недавно превратился в вирус, патогенный для человека и ондатр, в силу эффекта «бутылочного горлышка» все патогенные штаммы имели бы общего предка, возникшего в 30-40-х годах прошлого века. Наши датировки противоречат этой гипотезе. Примечательно, что 2 штамма ВОГЛ, изолированных от больных, располагаются на отдаленных ветвях филогенетических деревьев (см. рисунок). Более вероятно, что ВОГЛ циркулировал в Сибири в течение, как минимум, последнего тысячелетия и использовал интродуцированный высокочувствительный вид O. zibethicus, как «амплифицирующего хозяина», что привело к росту популяции вируса, появлению дополнительных путей передачи ВОГЛ человеку и росту заболеваемости ОГЛ. С этим предположением согласуется и тот факт, что не выявлено единого места происхождения всех существующих в настоящее время генотипов ВОГЛ, что указывает на достаточно длительную эволюцию вируса именно в Сибири. Таким образом, современный филогенетический и филогеографический анализ подтверждает предположение об автохтонном происхождении ВОГЛ, впервые высказанное еще в 1966 г. [34].
Наше исследование, показывая обмен штаммами ВОГЛ между эндемичными областями, ставит также вопрос: что удерживает ВОГЛ в пределах его нынешнего ареала, если его переносчики и хозяева, например, клещи D. reticulatus, полевки A. terrestris, и ондатры, распространены в ХХ веке практически по всей Северной Евразии в сходных климатических и экологических условиях? В отличие от ВОГЛ, ареалы многих (хотя и не всех) флавивирусов, переносимые клещами, занимают почти всю экологически благоприятную территорию, причем зафиксировано достаточно быстрое перемещение видов, генотипов и штаммов таких вирусов на большие расстояния [32, 35, 36]. Возможно, ключевой элемент эпидемиологии ОГЛ – специфический вид резервуарных хозяев или «сверхкомпетентный» переносчик, распространенный только в Западной Сибири, – ещё не обнаружен.
В заключение отметим, что решение поставленной задачи методологически состояло из нескольких этапов, каждый из которых обладал собственной спецификой и соответствующим математическим инструментарием. На первом этапе была показана принципиальная возможность реконструкции эволюции ВОГЛ на основе имеющихся данных о нуклеотидных последовательностях 25 штаммов ВОГЛ. Априори это не было очевидно, поскольку технически моделирование было бы невозможно, если бы анализируемые последовательности были слишком короткими или слишком сходными, если бы число изученных штаммов было слишком мало или они не различались бы существенно по времени и месту изоляции и т. п. На втором этапе из множества теоретически возможных математических моделей эволюции ВОГЛ была выбрана конкретная модель, наиболее адекватно описывающая имеющийся массив данных. На третьем этапе было проведено собственно моделирование, по результатам которого были определены топология и параметры Байесовского датированного филогенетического дерева и Байесовского филогеографического дерева.
* * *
Выражение признательности
Определение нуклеотидных последовательностей штаммов ВОГЛ выполнено при финансовой поддержке Международного Научно-технического Центра, грант № 2087 «Разработка и внедрение методов борьбы с новыми и вновь возникающими арбовирусными инфекциями в России и США с особым вниманием на энцефалит Западного Нила».
