
В России за последние 20 лет количество дженериков росло лавинообразно, и в 2013 г. их доля на рынке составляла 77%. Это часть общемирового процесса: по мере того как истекают патенты на оригинальные лекарства, появляется все больше дженериков. Несмотря на очевидные экономические преимущества воспроизведенных лекарств, они часто отличаются от оригинального препарата по профилю эффективности и безопасности. Чтобы понять, не принесет ли дженерик больше вреда, чем пользы, необходимо исследовать его биоэквивалентность. В мировой практике нет единого стандарта экспертизы биоэквивалентности [1]. Каждая страна решает этот вопрос, исходя из особенностей своих законов. Очень часто возникают юридические разночтения. В частности, многие страны по-разному толкуют понятие «дженерик», и поэтому Всемирная организация здравоохранения (ВОЗ) рекомендует использовать термин «мультиисточниковые лекарственные препараты» (англ. multi-source pharmaceutical products) [2]. Это фармацевтически эквивалентные или фармацевтически альтернативные препараты, которые могут быть или не быть терапевтически эквивалентными.
Эта формулировка допускает широкое толкование. Препараты могут быть признаны фармацевтическими эквивалентами, если они содержат одинаковую фармацевтическую субстанцию в той же лекарственной форме, с тем же путем введения и совпадают по дозировке или концентрации. При этом они могут различаться по форме, конфигурации риски, механизму высвобождения, сроку годности и вспомогательным веществам. Следовательно, если свойства препарата зависят от вспомогательных веществ, то у дженерика они будут другими, даже если он признан фармацевтическим эквивалентом.
В США приняты более строгие правила. В Оранжевой книге (Orange Book), основном справочнике по биоэквивалентности, специально сказано, что «лекарственные продукты считаются терапевтическими эквивалентами, только если они являются фармацевтическими эквивалентами, а также если от них ожидается тот же клинический эффект и профиль безопасности…». Следовательно, согласно требованиям Управления по контролю за пищевыми продуктами и лекарственными препаратами США (Food and Drug Administration – FDA), дженерик может заменить оригинальный препарат, только если он признан терапевтически эквивалентным [3].
 Представления о терапевтической и биоэквивалентности
Представления о терапевтической и биоэквивалентности
Биоэквивалентность не означает автоматически терапевтической эквивалентности, точно так же, как успехи препарата в лаборатории не всегда переходят в клиническую практику. Под биоэквивалентными лекарственными препаратами понимаются фармацевтически эквивалентные или фармацевтически альтернативные препараты, которые показывают сопоставимую биодоступность. Есть несколько подходов к определению биоэквивалентности. Например, 2 препарата будут биоэквивалентны, если исследуемый препарат по скорости и степени абсорбции несущественно отличается от скорости и степени абсорбции референтного препарата при однократном или многократном их применении в одинаковой молярной дозе фармацевтической субстанции в аналогичных экспериментальных условиях.
Биоэквивалентность – это одно из требований для терапевтической эквивалентности (взаимозаменяемости). Согласно подходам FDA и Европейского агентства по лекарственным средствам (EMA), 2 препарата могут быть признаны терапевтическими эквивалентами, если:
1. Они являются фармацевтическими эквивалентами в силу:
- идентичного содержания одинаковой фармацевтической субстанции в одинаковой лекарственной форме при одинаковом пути введения и
- соответствия фармакопейным или иным действующим стандартам по дозировке, качеству, чистоте и подлинности.
2. Они являются биоэквивалентными, то есть:
- в отношении них отсутствуют известные или потенциальные причины небиоэквивалентности (например, водные растворы для внутривенного введения заведомо признаются биоэквивалентными), и они удовлетворяют приемлемым стандартам in vitro (биовейвер) или
- при наличии таких известных или потенциальных причин они удовлетворяют надлежащим стандартам биоэквивалентности (то есть проведены исследования биэквивалентности).
3. Подтверждены их безопасность и эффективность.
4. Они сопровождаются правильной (обоснованной) информацией о лекарственном препарате в его инструкции по применению.
5. Они производятся в соответствии с текущими правилами надлежащей производственной практики.
Этот перечень устанавливает строгие требования, потому что, пропустив на рынок дженерики низкого качества, государство рискует понести большие расходы из-за последствий неэффективного лечения.
В России, согласно Федеральному закону от 22.12.2014 № 429-ФЗ «О внесении изменений в Федеральный закон „Об обращении лекарственных средств”» [4], в статье 27.1 утвержден порядок определения взаимозаменяемости лекарственных препаратов для медицинского применения. Взаимозаменяемость лекарственных препаратов определяется на основе следующих параметров:
- эквивалентность качественных и количественных характеристик фармацевтических субстанций;
- эквивалентность лекарственной формы, то есть одинаковость способа введения и способа применения, а также сопоставимость фармакокинетических характеристик фармакологического действия, способность обеспечить достижение клинического эффекта;
- эквивалентность состава вспомогательных веществ (различия допустимы, если при проведении исследования биоэквивалентности доказано отсутствие клинически значимых различий фармакокинетики, безопасности и эффективности);
- идентичность способа введения и применения;
- отсутствие клинически значимых различий при проведении исследований биоэквивалентности или, в случае невозможности проведения этого исследования, отсутствие клинически значимых различий безопасности и эффективности лекарственного препарата при исследовании терапевтической эквивалентности;
- соответствие производителя лекарственного средства требованиям надлежащей производственной практики (GMP).
Согласно закону, терапевтическая эквивалентность – это «достижение клинически сопоставимого терапевтического эффекта при применении лекарственных препаратов для медицинского применения для одной и той же группы больных по одним и тем же показаниям к применению». Взаимозаменяемый лекарственный препарат – «лекарственный препарат с доказанной терапевтической эквивалентностью или биоэквивалентностью в отношении референтного лекарственного препарата, имеющий эквивалентные ему качественный состав и количественный состав действующих веществ, состав вспомогательных веществ, лекарственную форму и способ введения». В качестве референтного (оригинального) препарата признается «лекарственный препарат, который впервые зарегистрирован в Российской Федерации».
Производители дженериков (воспроизведенных лекарственных препаратов) должны доказывать их взаимозаменяемость. Отдельного внимания требуют вспомогательные вещества, потому что от их подбора часто зависит эффективность лекарственного средства.
Роль вспомогательных веществ в технологии изготовления лекарственных средств. Технологии повышения биодоступности
Фармацевтические компании ежегодно скринируют тысячи молекул. Многие из них плохо растворяются в воде, поэтому на их основе сложно сделать лекарство с хорошей биодоступностью. Чтобы спрогнозировать биодоступность, разработана биофармацевтическая классификация лекарств (BCS). Она учитывает параметры растворимости действующего вещества и проницаемости через стенки желудочно-кишечного тракта (ЖКТ). Для сравнения используется биодоступность при внутривенной инъекции, которая принимается за 100%. Если примерно 90% дозы попадает в кровь при приеме реr os, то биодоступность считается высокой. Выделяют четыре класса биодоступности, причем II и IV классы представляют особые трудности [5]
Что делать, если действующее вещество обладает низкой растворимостью и плохой всасываемостью? Можно увеличить дозу, но это не всегда эффективно. Если биодоступность конкретной молекулы составляет всего несколько процентов, то чтобы обеспечить необходимую концентрацию в крови, потребуется очень большая доза. Пациенту придется принимать по несколько таблеток 3 или 4 раза в день. Это скажется на частоте побочных эффектов и приверженности к терапии. С такой проблемой врачи часто сталкивались, назначая первые антиретровирусные препараты. Можно изменить и способ доставки, однако большинство препаратов не подходят для чрескожного или ингаляционного введения, а ежедневные внутривенные инъекции технически недоступны и грозят осложнениями.
Повысить биодоступность действующего вещества при пероральном приеме можно методами современной фармакологии. Для этого необходимо, чтобы концентрация лекарства в ЖКТ была достаточно высокой и сохранялась достаточно долго. Когда пациент принимает таблетку, содержащую действующее вещество в кристаллической форме, оно растворяется, пока концентрация раствора не достигает термодинамической стабильности, после чего остается на этом уровне. Если этой концентрации не хватает для адекватной биодоступности, можно создать таблетку с действующими веществом в аморфном состоянии. Это означает, что кристаллическая структура вещества разрушается, и молекулам проще освободиться. Когда пациент принимает такую таблетку, концентрация действующего вещества в ЖКТ повышается гораздо быстрее. В специальной литературе это явление называют «эффектом пружины» (spring effect) [6]. За счет повышенной концентрации биодоступность начинает расти. Однако это длится недолго, потому что в перенасыщенном растворе снова начинают формироваться кристаллы, которые затрудняют всасывание.
Чтобы сохранить концентрацию на нужном уровне, необходимо замедлить кристаллизацию, внедрив в таблетку полимерный носитель. Тогда концентрация снижается медленно, и действующее вещество может проникнуть в кровоток в необходимой дозе. Постепенное снижение концентрации действующего вещества называют «эффектом парашюта». Правильно подобранный полимер поможет сначала быстро достичь высокой концентрации, а потом поддерживать её достаточно долго. Поскольку кристаллизация аморфного вещества может происходить и просто при хранении таблетки, полимер необходим, чтобы повысить сроки годности, особенно при перепадах температуры и высокой влажности. Универсальных рецептов здесь нет, для каждого действующего вещества подбирают подходящий именно этой молекуле полимер-носитель [6].
Подобрав полимер, нельзя просто перемешать его с действующим веществом. Их нужно растворить друг в друге. Однако полученный раствор в жидком виде недолго сохранит свои свойства. Чтобы добиться стабильности, необходимо создать твердый раствор аморфного действующего вещества в полимере. Для этого существует несколько технологий: экструзия расплава, сушка распылением, испарения растворителя и другие. Например, экструзия расплава подразумевает, что действующее вещество сначала растворяют в термопластичной матрице, состоящей из полимера и других действующих веществ. Полученный раствор пропускают через специальный аппарат – экструдер и получают твердый стекловидный раствор. Его пластифицируют, объединяют с другими вспомогательными вещества и прессуют в таблетки. Затем таблетки покрывают оболочкой, придавая им нужную форму и цвет [7].
Чтобы получить лекарство с высокой биодоступностью, необходимо создать новую, уникальную линию производства. Поэтому когда разработчики дженериков пробуют воспроизвести оригинальный препарат, им необходимо не просто скопировать состав, а построить аналогичную линию. Чтобы оценить успех этой работы, нужно изучить биоэквивалентность дженерика в сравнении с оригинальным препаратом.
Требования к носителю
Твердые растворы (твердые дисперсии) действующих веществ и вспомогательных веществ – один из методов повышения растворимости. Вспомогательные вещества становятся основой для твердого раствора. Они позволяют изменить физическое состояние действующего вещества с кристаллического на аморфное, повышая тем самым его растворимость. В аморфном состоянии вещества молекулы расположены по отношению друг к другу менее упорядоченно, потому его высвобождение происходит гораздо быстрее, обеспечивая нужную концентрацию [8]. Полимер для твердого раствора должен быть:
- токсикологически приемлемым для перорального приема;
- химически и физически совместимым с действующим веществом;
- подходящим для дальнейшего прессования в таблетки;
- растворимым в водной среде с рH от 1 до 7;
- способным подавить рекристаллизацию после растворения.
Каждая молекула требует своего сочетания вспомогательных веществ. Просчет в подборе может привести к тому, что действующее вещество так и не станет растворимым или быстро растворится, а потом также быстро рекристаллизуется.
Истории оригинальных препаратов и дженериков
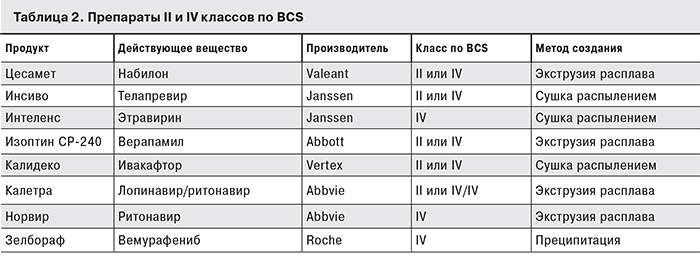
Когда речь идет о лекарственных препаратах II и IV классов по BCS, в каждом случае необходимо подробно изучить историю создания этого препарата. Практикующим врачам и пациентам это поможет оценить усилия фармацевтических компаний, а производителям дженериков – более эффективно воспроизвести технологию. В табл. 2 перечислены некоторые из таких препаратов [9].
Такролимус (Prograf)
Биоэквивалентность не обязательно говорит о терапевтической эквивалентности. Обычно биоэквивалентность изучают у здоровых субъектов, однако у больных с пересаженной почкой метаболизм и экскреторные функции работают иначе. Во многом успех трансплантации зависит от эффективности иммуносуппресоров. Препарат такролимус используют для предотвращения отторжения пересаженных тканей. Существует несколько дженериков такролимуса, которые широко применяют в странах третьего мира.
В 2008 г. J.A. Petan и соавт. [10] опубликовали работу, в которой сравнили физико-химические свойства оригинального такролимуса (Prograf) и пяти его дженериков производства компаний из Мексики, Чили, Индии и Южной Кореи. Скорость растворения всех пяти дженериков отличалась от скорости растворения оригинального препарата. Tacrobell и T-Inmun растворялись быстрее, чем Prograf, а Tenacrine, Framebin и Talgraf, напротив, растворялись медленно и не полностью. За 2 ч они выпустили от 24 до 51% действующего вещества. В клинической практике подбор дозы необходим, чтобы поддерживать концентрацию препарата в заданных пределах. При избытке такролимуса в крови могут развиться побочные эффекты, в том числе нефро- и нейротоксические, при нехватке может произойти отторжение трансплантата. Препарат с непредсказуемой растворимостью, в котором к тому же содержится неизвестное количество действующего вещества, опасен для пациентов [11].
Различия между оригинальным такролимусом и дженериками может быть связано с особенностями производства препарата. Такролимус – это вещество с низкой растворимостью (II класс по BCS). Чтобы улучшить его растворимость, K. Yamashita и соавт. [12] провели серию экспериментов, выбрали лучший полимер-носитель и разработали новый способ создания твердого раствора. Вероятно, именно эту авторскую технологию не смогли воссоздать производители дженериков.
Можно ли создать эффективный дженерик такролимуса? Конечно, да, ведь при соблюдении технологии получившийся препарат обязательно будет соответствовать критериями биоэквивалентности. Вопрос заключается в том, насколько производители дженериков владеют технологией. В случае с каждым препаратом II или IV классов по BCS требуется не только следование описанным шагам и закупка указанных в лицензии компонентов. Чтобы твердая дисперсия действующего вещества стала эффективным лекарством, требуется индивидуальная «доводка» технологического процесса. Множество настроек аппаратуры, тонкости хранения материалов, нюансы композиций – все это невозможно просто скопировать.
Лопинавир/ритонавир (LPV/r, Kaletra)
Калетра является антиретровирусным препаратом из группы ингибиторов протеазы. Препарат был изначально зарегистрирован FDA в виде желатиновых капсул. Это помогало отчасти решить проблему низкой растворимости лопинавира и ритоновира. Эти капсулы нужно было хранить в холодильнике, потому что даже при комнатной температуре композиция теряла стабильность. Чтобы оптимизировать процесс всасывания препарата, следовало принимать его строго после еды по 6 капсул в сутки. Все это порождало неудобства для пациентов и негативно отражалось на приверженности к терапии. Чтобы преодолеть эти трудности, разработчики решили создать твердый раствор [13].
Прежде всего следовало определить, каким должно быть соотношение лопинавира, ритонавира и вспомогательных веществ. Первая комбинация оказалась неудачной, и тогда были разработаны еще 5 комбинаций, которые отличались по долям действующих веществ, процедуре формовки, типу поверхностно-активного вещества и другим параметрам. Сравнив их между собой, авторы выбрали 3 наиболее эффективных и сравнили их уже с желатиновыми капсулами. Одна из комбинаций оказалась независимой от приема пищи и более простой в изготовлении. Последовала серия модификаций: изменили содержание красителей, чтобы достичь желаемого цвета, и заменили 2 вспомогательных вещества. Убедившись в адекватной биодоступности новых составов, разработчики создали пробную линию для получения таблеток в производственном объеме. Получившиеся таблетки снова сравнили с капсулами. Наконец, было проведено второе исследование биодоступности, в котором 3 разных серии таблеток показали соответствие критериям биоэквивалентности относительно референтного препарата. Чтобы оценить стабильность новой формы, таблетки размещали на длительное хранение в разных условиях, а также подвергали стресс-тестированию. За исключением нескольких таблеток, деформированных в условиях высокой влажности, препарат сохранил свои свойства в течение 18 мес. [14].
LPV/r – один из первых препаратов IV класса по BCS, на основе которого был сделан твердый раствор. Разработка патентованной технологии потребовала от компании 10 лет работы и вложений. Поскольку спрос на ингибиторы протеазы остается высоким, ряд компаний попытались повторить этот опыт. На сегодняшний день исследований эффективности этих препаратов немного. В частности, один из дженериков LPV/r продемонстрировал адекватные уровни действующего вещества в плазме и хорошую переносимость у больных ВИЧ-инфекцией [15]. В то же время несколько дженериков LPV/r в исследованиях биодоступности на собаках показали низкие результаты, вплоть до 1% [16].
В чем причина неудачи дженерика, если его состав идентичен оригинальному препарату? Вероятнее всего, вспомогательные вещества не смогли обеспечить достаточно быстрого высвобождения действующего вещества или замедлить процесс рекристаллизации раствора. С клинической точки зрения эти отличия означают, что при приеме дженерика концентрация действующих веществ в крови окажется заведомо меньше. На этом фоне есть риск развития резистентности или вирусологической неудачи [17].
Создавая фармацевтическую композицию из двух действующих веществ и нескольких вспомогательных, необходимо постоянно проверять их биодоступность и биоэквивалентность. В идеале это нужно делать на каждом шагу, причем не только в тех случаях, когда речь идет о составе препарата, но и когда проводится отладка производства.
Этравирин (Intelence)
Интеленс (этравирин) относится к группе ненуклеозидных ингибиторов обратной транскриптазы (ННИОТ) ВИЧ-1. Этот фермент отвечает за транскрипцию вирусной РНК в ДНК, которая потом интегрируется в геном клетки. На фоне приема других ингибиторов ННИОТ (невирапина, эфавиренза и делавирдина) нередко развиваются мутации, в частности K130N и Y181C. В процессе поиска веществ, эффективных против этих штаммов, и был обнаружен этравирин. Он применяется исключительно у взрослых, обычно уже получавших терапию, испытавших вирусологическую неудачу и имеющих признаки резистентности к ННИОТ и другим антиретровирусным препаратам [18].
Этравирин относится к IV классу по BCS. Изначально разработчики создали капсулы на основе полиэтиленгликоля. Однако этот носитель оказался недостаточно эффективным, и его заменили на метилоксипропилцеллюлозу. В этой форме препарат прошел исследования II фазы, однако чтобы обеспечить оптимальную биодоступность, разработчики решили использовать другой метод и остановили свой выбор на сушке распылением. В этой форме препарат оказался существенно эффективнее предыдущих вариантов и прошел путь от III фазы до фармацевтического рынка. В процессе разработки было проведено несколько исследований биоэквивалентности. Третья форма этравирина в дозе 200 мг 2 раза в сутки обеспечила большую экспозицию, чем предыдущая в дозе 800 мг 2 раза в сутки [19].
Этравирин – это пример того, что, вопреки сложившемуся заблуждению, биоэквивалентность необходимо изучать не только в рамках оценки дженериков. Корректные критерии биоэквивалентности сокращают время разработки новых лекарственных препаратов. Исследования этравирина продолжаются. Некоторые авторы [20] экспериментирует с разными полимерами и заявляют об увеличении его растворимости in vitro от 4 до 15 раз. Прежде чем результаты этих опытов будут применены в клинике, также необходимо изучить биоэквивалентность новых составов.
Качество лекарственного средства определяет качество лечения
В отличие от многих других коммерческих продуктов невозможно оценить качество лекарства, просто посмотрев на него. Пациентам и специалистам необходимы сведения об эффективности и безопасности лекарственных средств. Сэкономив на оригинальном препарате, можно столкнуться с последствиями неэффективного лечения. Это особенно актуально для препаратов II и IV классов по BCS. Этих лекарств не так много. Каждое из них – пример успешного решения сложной технологической задачи. Часто они используются для лечения больных с тяжелыми социально-значимыми хроническими заболеваниями. Если такой препарат, например, антиретровирусный, окажется неэффективным, то возникшая резистентность станет угрозой для пациента и популяции в целом. Когда производитель дженерика изменяет композиционный состав вспомогательных веществ и технологию производства, необходимо особенно тщательно проверить результаты его работы. Таким экзаменом должны стать исследования терапевтической эквивалентности и биоэквивалентности.


